謬誤,目前空調是當前制冷方法中最科學最節能的,具體原因可以往下看看。
1、瓦斯狀態方程式
先認識3個概念:
a)構成物質分子聚合狀態稱為物質的 聚集態 :氣態、液態、固態、電漿態、超密態、BEC等;(液固為凝聚態)
b)沒有外力作用,物理、化學性質完全相同,成分完全相同的均勻物質聚集態,稱為 相;
c)處於平衡態的某種物質的熱力學參量之間所滿足的函式關系稱為該物質的 狀態方程式 。
1.1理想瓦斯
在壓力極低時,分子間的距離非常大,此分時子間的交互作用非常小,而分子本身線度與分子間的距離相比可忽略不計,因此可將分子看作是沒有體積的質點,這種狀態下的瓦斯可近似看作理想瓦斯,理想瓦斯在微觀上具有以下兩個特征:
①分子間無交互作用力;②分子本身不占有體積因此只有壓力趨於零時的瓦斯才是理想瓦斯,理想瓦斯可看作是真實瓦斯在壓力趨於零時的極限情況。對於理論上的理想瓦斯 在任何溫度、任何壓力下均服從理想瓦斯狀態方程式 。
玻意耳(Boyle, 1627-1691)定律: 物量和溫度恒定時,體積與壓力成反比
pV=C (1-1)蓋·呂薩克(Gay Lussac, 1778-1850)定律: 物量與壓力恒定時,體積與熱力學溫度成正比
V=V_{0}(1+\beta_{p}t) (1-2)
對於等壓體膨脹系數,表示壓力不變時單位溫度所引起的體積變化
\beta_{p}=\frac{1}{V}(\frac{\partial V}{\partial T})_{p} (1-3)
式中 V 和 V_{0} 表示溫度為 t 和 0℃ 的體積, \beta_{p}=1/273.15 ℃ ,熱力學溫度 T=(273.15+t/ ℃)K ,因此 V=V_{0}\beta_{p}T
\frac{V_{2}}{{V_{1}}}=\frac{T_{2}}{T_{1}} (1-4)查理(Charles, 1746-1823)定律: 物量與體積不變時,壓力與熱力學溫度成正比
p=p_{0}(1+\alpha_{V}t) (1-5)
對於壓力系數,表示體積不變時單位溫度所引起的壓力變化
\alpha_{V}=\frac{1}{p}(\frac{\partial p}{\partial T})_{V} (1-6)
式中 p 和 p_{0} 表示溫度為 t 和 0℃ 的壓力, \alpha_{V}=1/273.15 ℃ ,熱力學溫度 T=(273.15+t/ ℃)K ,因此 p=p_{0}\alpha_{V}T
\frac{p_{2}}{{p_{1}}}=\frac{T_{2}}{T_{1}} (1-7)以上實驗得出的三個定律,歸納後可得出理想瓦斯狀態方程式
pV=nRT (1-8)上式中 n 為物量,有些物理教材會用 \nu 代表。對於標準狀況下 T=273.15K , p=1.013\times 10^{5}N\cdot m^{-2} , V_{m}=22.4\times 10^{-3}\ m^{3} ,帶入上式得 普適瓦斯常數
R=8.31J\cdot mol^{-1}\cdot K^{-1}對於混合理想瓦斯,根據道爾頓定律: 混合瓦斯的總壓力等於各組分單獨存在於混合瓦斯的溫度、體積條件下所產生壓力的總和
p=\sum_{B}{p_{B}}=\sum_{B}\frac{n_{B}RT}{V} (1-9)
1.2真實瓦斯
理想瓦斯分子沒有交互作用,所以在任何溫度、壓力下都不會液化。但真實瓦斯分子間交互作用勢能隨分子間距離的變化類似重力勢能變化。降低溫度與增加壓力可使瓦斯的莫耳體積減小,即 分子間距離減少,這將使分子間相互吸重力作用增加,導致瓦斯變成液體 。
在某一適當溫度下,液體與其蒸汽可達成一種動態平衡,即單位時間內由液體分子變為瓦斯分子的數目與由瓦斯分子變為液體分子的數目相同,也就是說液體的蒸發速度與瓦斯的液化速度相同,此時這種氣-液平衡時的瓦斯稱為飽和蒸汽,所具有的壓力稱為 飽和蒸氣壓 p^{*} 。當飽和蒸氣壓與外界壓力相等時,液體沸騰,此時相應的溫度稱為液體的 沸點 。我們先來看看3種常見物質的飽和蒸氣壓
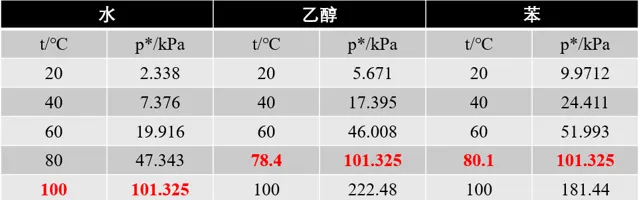
圖中標紅的數據為標準大氣壓下的正常沸點,根據道爾頓定律,此時無論空氣中該物質組分所占含量,即便空氣中水蒸汽含量達到100%,但已達到飽和蒸氣壓,液體表面的分子會發生汽化,內部的分子也會汽化。在一定溫度下如果蒸汽分壓小於飽和蒸氣壓,液體將蒸發,直到蒸發至蒸汽分壓升至飽和蒸氣壓;反之,如果蒸汽分壓大於飽和蒸氣壓,瓦斯將液化,直到液化至蒸汽分壓降至飽和蒸氣壓。例如,水在20℃時飽和蒸氣壓2.338kPa,但只要水蒸氣的分壓小於2.338kPa,液體就會蒸發,這就是衣服能在常溫下自然晾幹的原因;夜晚降溫時,飽和蒸氣壓降低,空氣中水蒸汽分壓大於飽和蒸氣壓,因此液化出現露珠。
液體的飽和蒸氣壓隨溫度的升高而增大,從理想瓦斯狀態方程式也可以看出,溫度升高若要使體積不變,則壓力越大。但對於真實瓦斯,有一個特殊的溫度, 在該溫度以上,無論施加多大壓力,都不再能使瓦斯液化 ,這個溫度稱為 臨界溫度 T_{c} ,臨界溫度是瓦斯能夠液化所允許的最高溫度。
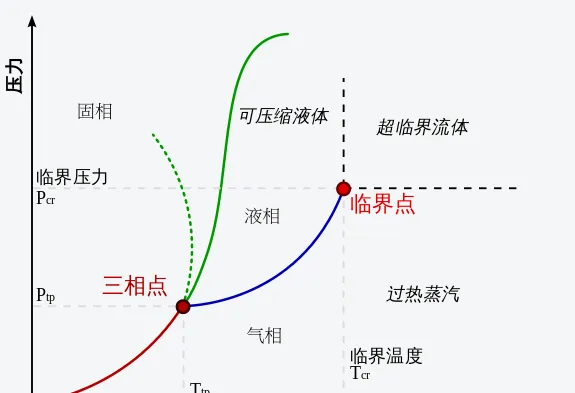
給定一種組成,透過實驗可以發現在特定溫度及壓力下只有某些相可以存在,把這些相在不同壓力和溫度時的實驗結果繪制成 相圖 。在臨界溫度 T_{c} 時對應的飽和蒸氣壓稱為 臨界壓力 p_{c} ,也就是與相圖中液氣共存線段的交點,稱為 臨界點 。當溫度及壓力接近臨界點時,液相和氣相的性質會越來接近,在臨界點時無法區分液相和氣相,溫度及壓力一旦超過臨界點,就沒有單獨可區分的液相或氣相,只會有一種稱為超臨界流體的流體相。超臨界流體可以理解為一種稠密的氣相,其具有瓦斯的黏度與液體的密度,擴散系數比液體大得多,再加大壓力就不屬於工程上研究的問題了。


在相圖中我們還可以看到左下方有一個使一種物質三相達到熱力學平衡共存時的一組溫度和壓力數值,這就是 三相點 ,在這個溫度和壓力附近,一點點擾動就會使物質發生沸騰或凝固,變得非常不安定。比如,水 的固-液-氣-三相點是0.01℃ (273.16K )及611.73Pa (約等於標準大氣壓101.325kPa 的千分之六)。在上面的動圖中我們可以看出三相點左下方,物質是可以輕易發生氣-固轉變的,比如沸騰的氣泡突然凝固,凍結的固體突然漲裂。反之,現實中有些自然昇華和凝華的物質,其實在高溫高壓下也是有液體狀態的。
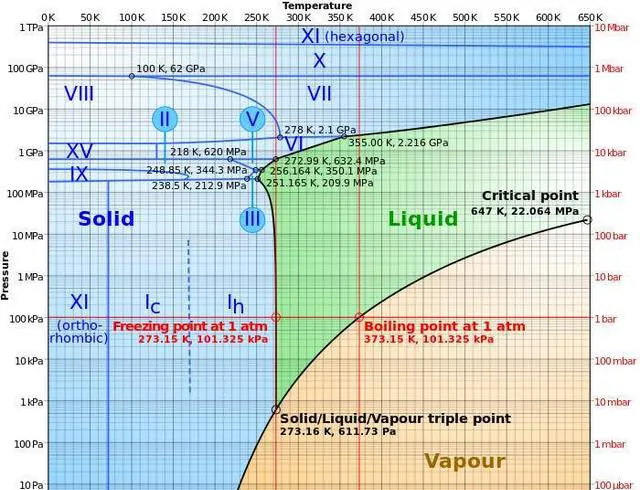
上面僅僅針對的是水而言,但是如果物質有多種固相和液相,測相圖上相鄰的相之間都會形成三相點。許多固體(如鐵、冰)在不同溫度和壓力下會有不同的固相,因此有多個三相點。如冰有正交晶系和六方晶系等9種晶體結構,鐵在冷卻過程中會經歷 \delta 體心立方、 \gamma 面心立方、 \beta 體心立方、 \alpha 體心立方相變,如果是兩相共存還要參考鐵碳相圖,過程會更加復雜。不過,氦 -4是唯一有兩種液相(低溫下液氦發生二級相變 )的物質,所以氦-4雖然沒有固-液-氣-三相點,但有液-液-氣-三相點:2.177 K, 5.043 kPa。
範德華(van der Waals) 認為理想瓦斯的壓力是指分子間無交互作用力時的壓力,莫耳體積是每莫耳瓦斯分子自由活動的空間;而真實瓦斯處在實際的p,Vm,T條件時,由於分子間有交互作用,壓力p實際上是理想瓦斯在同等T,Vm時所應有的壓力減去由於內部吸重力造成的壓力減小後的結果,即 p=p_{理}-a/V_{m}^{2} ;而體積在考慮了分子本身占有的體積b後,自由活動的空間 V_{m自由}=V_{m}-b ,因此將修正後的壓力和體積帶入理想瓦斯狀態方程式
(p+\frac{a}{V_{m}^{2}})(V_{m}-b)=RT (1-10)上式為範德華方程式,將 V_{m}=V/n 代入上式
(p+\frac{an^2}{V_{}^{2}})(V_{}-nb)=nRT (1-11)得到適用於其物量為n的範德華方程式,式中的a,b為範德華常數。經過範德華修正後,真實瓦斯也有自己的狀態方程式了。

1.3微觀模型
熱力學第零定律 (Zeroth Law of Thermodynamics)也稱為熱平衡定律:
若兩個熱力學系統均與第三個系統處於熱平衡狀態,此兩個系統也必互相處於熱平衡。說明白一點就是熱0給出了溫度的概念(互為熱平衡的物體間溫度相同),因此建立了共同的熱力學溫標。
白努利(Daniel Bernoulli, 1700—1782)設想瓦斯壓力來自例子碰撞器壁所產生的沖量。任何宏觀可測定量均是某微觀量的平均統計值,所以器壁所受到的瓦斯壓力是單位時間內大數分子頻繁碰撞器壁所給予單位面積器壁的平均總沖量。這種碰撞是無間歇的,所施加的力也是恒定不變的,在這裏給出微觀瓦斯壓力公式,n為分子數量
p=\frac{1}{6}n\tilde{v}\cdot2m\tilde{v}\approx\frac{1}{3}nm\tilde{v^{2}} (1-12)同樣可以計算出分子的平均動能
\bar{\varepsilon_{t}}=\frac{1}{2}m\bar{v^{2}} (1-13)值得一提的是,瓦斯壓力不僅存在於器壁,也存在於瓦斯內部,對於理想瓦斯,這兩種壓力運算式完全相同。我們將微觀量與宏觀量進行聯系,可得到 波茲曼常數k
k=\frac{R}{N_{A}}=1.38\times10^{-23}[J\cdot K^{-1}]
最後將微觀與宏觀運算式進行聯系p=nkT,可得 單原子 分子熱運動平均動能
\bar{\varepsilon_{t}}=\frac{3}{2}kT (1-14)莫耳內能則為
U_{m}=N_{A}\cdot\frac{3}{2}kT=\frac{3}{2}RT (1-15)上式表明分子熱運動平均平動動能與絕對溫度成正比,絕對溫度越高,分子熱運動越劇烈。絕對溫度是分子熱運動劇烈程度的量度, 粒子的平均熱運動動能與粒子品質無關,而僅與溫度有關 。 \bar{\varepsilon_{t}} 是分子雜亂無章熱運動平均平動動能(內能),它不包括整體定向運動動能(機械能)。
2、功與熱,焓與熵
先記住3個定義:
a)熱力學中對於功與熱這種 過程量 取變分而不取微分,如 đW、đQ(物理化學教材裏用δW、δQ),而對於內能、焓、熵這種 狀態量 取微分而不取變分,如 dU、dH、dS,我們可以不用糾結數學上的定義,在這裏只是借用兩種符號區別一下這幾種因次[J]相同的物理量。
b)只有系統內部各部份之間及系統與外界之間都始終同時滿足力學、熱學、化學平衡條件的過程才是 準靜態過程 。
c)只有無耗散的準靜態過程才是 可逆過程 。
2.1熱力學第一定律
功是在力學交互作用過程中系統和外界之間轉移的能量 。熱力學中的力學交互作用力是一種廣義力,它不僅包括機械力(壓力、拉力、表面張力等),也包括電場力、磁場力等,所以功也是一種廣義功,這裏有4點需要註意:
1)只有在系統狀態變化過程中才有能量轉移,系統處於平衡態時能量不變,因而沒有做功, 功與系統狀態無對應關系,說明功不是狀態量 ;
2)只有在廣義功作用下產生了廣義位移後才做了功;(如壓力、電動勢在體積變化和電荷量遷移後)
3)非準靜態過程中,由於系統內部壓力處處不同,且隨時在變化,很難計算系統對外做的功,因此 系統對外做功計算局限於準靜態過程 ;
4)外界對瓦斯做 功 以 W 表示,瓦斯對外界做功以-W表示。
瓦斯對外界做功以體積膨脹形式進行,此時對瓦斯分析有đW=-pdV,該過程是無限小的可逆過程中外界對瓦斯所做元功的運算式,它是系統狀態量p、V的函式。đW>0,表示外界對系統做正功,此時dV<0表示瓦斯被壓縮,反之則膨脹。在整個可逆過程中有
W=-\int_{V_{1}}^{V_{2}}pdV (2-1)表示外界對系統做的總功。上述過程存在的問題是同時存在2個自變量T=T(p,V),實際上並不用重積分,只需令每一種準靜態變化過程都對應於p-V圖中某一曲線,這時p與V就有一一對應關系。另外,在下圖中可以發現沿CD等溫線,沿CAD先等壓再等體,沿CBD先等體再等壓,三條曲線下的面積均不相等,這說明功與變化的的路徑有關,功不是系統狀態內容,不是狀態函式,元功đW不滿足多元函式中全微分條件。 đW僅表示沿某一路徑的無窮小變化,因此在微分符號d上加一杠đ以示區分 。
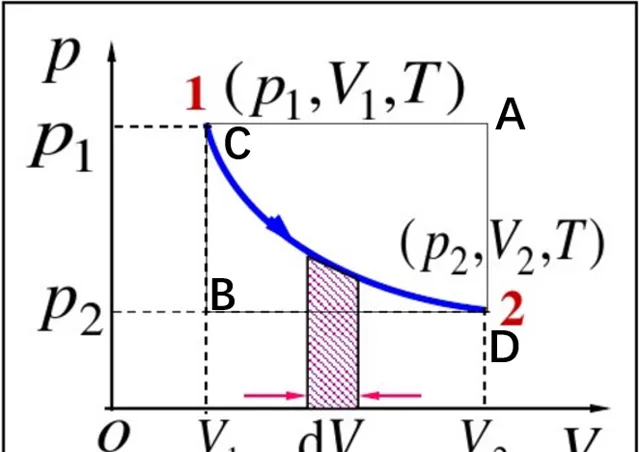
等溫過程:
W=-\int_{V_{1}}^{V_{2}}pdV=-nRT\int_{V_{1}}^{V_{2}}\frac{dV}{V}=-nRTln\frac{V_{2}}{V_{1}}=-nRTln\frac{p_{2}}{p_{1}} (2-2)等壓過程:
W=-\int_{V_{1}}^{V_{2}}pdV=-p\int_{V_{1}}^{V_{2}}dV=-p(V_{2}-V_{1}) (2-3)等體過程:
W=-\int_{V_{1}}^{V_{2}}pdV=0 (2-4)當系統狀態的改變來源於熱學平衡條件的破壞,也即來源於系統與外界間存在溫度差時,我們就稱系統與外界間存在熱學交互作用,有能量從高溫物體傳遞給低溫物體,這樣傳遞的能量稱為 熱量Q 。熱量和功是系統狀態變化中伴隨發生的兩種不同的能量傳遞形式,是不同形式能量傳遞的量度,它們都 與狀態變化的中間過程有關,因而不是系統狀態的函式 。
與功一樣,一個無窮小過程中所傳遞的熱量只能寫成đQ而不是dQ,因為不滿足多元函式全微分條件。功與熱量的區別在於它們分別來自不同的交互作用: 功由力學交互作用引起,只有產生廣義位移時才伴隨功的出現;熱量來源於熱學交互作用,只有存在溫差時才有熱量傳遞 。此外,熱力學中還有化學交互作用,擴散、滲透、相變、溶解稀釋、化學反應等都屬於這種。
熱力學第一定律 (First Law of Thermodynamics)其推廣和本質就是著名的 能量守恒定律 :
物體內能的增加等於物體吸收的熱量和對物體所做的功的總和。這裏的 內能U 是系統內部所有微觀粒子的微觀無序運動能以及總的交互作用勢能兩者之和。 內能是狀態函式 ,處於平衡態系統的內能是確定的,內能與系統狀態間有一一對應關系。熱1的數學運算式也就是 內能定理 ,內能的增加可來源於外界對系統做功W與從外界吸收的熱量Q
U_{2}-U_{1}=Q+W (2-5)對於無限小的過程,因為U是狀態函式,能滿足多元函式全微分條件
dU=đQ+đW (2-6)對於準靜態過程,結合(2-1)可得出
dU=đQ-pdV (2-7)如此一來,我們就把功、熱量、內能和理想瓦斯的狀態方程式聯系起來了。最後要註意一點 理想瓦斯 內能是溫度的函式U=U(T),與體積壓力無關。但對於 真實瓦斯U=(T,V),內能還是V的函式 ,所以瓦斯向真空自由膨脹時勢能增加(理想瓦斯無勢能),動能降低,溫度是會降低的。
dU=(\frac{\partial U}{\partial T})_{V}dT+(\frac{\partial U_{ }}{\partial V})_{T}dV (2-8)2.2熱力學第二定律
功和內能都能透過宏觀或微觀的數學運算式定義出來,對於同為過程量的熱量,我們可以用熱容進行描述。物體升高或降低 單位溫度 所吸收或放出的熱量稱為物體的 熱容C
C=đQ/dT (2-9)每莫耳物體的熱容稱為 莫耳熱容Cm ,有C=nCm單位品質物體的熱容稱為 比熱容c ,有C=mc。
在等體過程中,瓦斯與外界沒有功的交往,所吸收的熱量全部用來增加內能,因此 莫耳定體熱容 C_{V,m} 為
C_{V,m}=(\frac{\partial U_{m}}{\partial T})_{V} (2-10)在等壓過程中,吸收的熱量除用來增加內能外,還需使瓦斯膨脹對外做功,所以定壓比定體熱容大 。根據(2-7)可知đQ=dU+pdV(克勞修斯的熱1運算式),為了計算莫耳定壓熱容 C_{p,m} ,我們定義一個函式
H=U+pV (2-11)因為U、p、V都是狀態函式,故它們的組合 焓H 也是狀態函式,與內能和熱容類似,把h、Hm稱為比焓和莫耳焓,因此莫耳定壓熱容 C_{p,m} 為
C_{p,m}=(\frac{\partial H_{m}}{\partial T})_{p} (2-12)等壓過程中吸收的熱量等於焓的增量 。由於 C_{p,m} 和 C_{V,m} 的定義,可匯出兩者的關系
C_{p,m}-C_{V,m}=(\frac{\partial H_{m}}{\partial T})_{p}-(\frac{\partial U_{m}}{\partial T})_{V}=(\frac{\partial H_{m}+pV_{m}}{\partial T})_{p}-(\frac{\partial U_{m}}{\partial T})_{V}=(\frac{\partial U_{m}}{\partial T})_{p}+p(\frac{\partial V_{m}}{\partial T})_{p}-(\frac{\partial U_{m}}{\partial T})_{V}
根據(2-8) dU_m=(\frac{\partial U_m}{\partial T})_{V}dT+(\frac{\partial U_{ m}}{\partial V_m})_{T}dV_m ,在等壓時除以dT得
(\frac{\partial U_m}{\partial T})_{p}=(\frac{\partial U_m}{\partial T})_{V}+(\frac{\partial U_{ m}}{\partial V_m})_{T}(\frac{\partial V_m}{\partial T})_{p} ,代入 C_{p,m}-C_{V,m} 式中得
C_{p,m}-C_{V,m}=[(\frac{\partial U_{ m}}{\partial V_m})_{T}+p](\frac{\partial V_m}{\partial T})_{p} ,我們可以這樣看 (\frac{\partial U_{ m}}{\partial V_m})_{T}(\frac{\partial V_m}{\partial T})_{p} 相當於1mol物質等壓升溫1K時,體積膨脹克服分子間吸重力,從外界吸收的熱量, p(\frac{\partial V_m}{\partial T})_{p} 相當於體積膨脹對外界做功而吸收的熱量。
對理想瓦斯狀態方程式有 (\frac{\partial V_m}{\partial T})_{p}=\frac{R}{p} , (\frac{\partial U_{ m}}{\partial V_m})_{T}=0 ,代入上式,最終有邁耶公式
C_{p,m}-C_{V,m}=R (2-13)關於熱量的描述過程可以結合熱1運算式,đQ=n C_{V,m} dT+pdV,來描述等溫、等壓、等體過程
等溫過程: 理想瓦斯內能不變đQ=-đW=pdV
Q=-W=nRTln\frac{V_2}{V_1} (2-14)等壓過程: 吸收熱量為đQ=n C_{p,m} dT
Q=n\int_{T_{1}}^{T_{2}}C_{p,m}dT (2-15)等體過程: 系統對外界做功為零,吸收的熱量等於系統內能的增加đQ=n C_{V,m} dT
Q=n\int_{T_{1}}^{T_{2}}C_{V,m}dT (2-16)值得註意的是,等壓過程中其內能改變量與等體過程吸收熱量相同 U_1-U_2=n\int_{T_{1}}^{T_{2}}C_{V,m}dT 。同為過程量的功在等體過程可以不變化,那麽熱量也存在一種過程可以不變化,這就是絕熱過程。
絕熱過程: Q=0,系統絕熱膨脹對外做了多少功,內能就減少多少。由đQ=n C_{V,m} dT+pdV,可知
-pdV=n C_{V,m} dT (2-17)我們對理想瓦斯狀態方程式兩邊全微分pdV+Vdp=nRdT,有dT=(pdV+Vdp)/nR,代入(2-17),得 ( C_{V,m} +R)pdV=- C_{V,m} Vdp, 結合(2-13),並定義 \gamma=\frac{C_{p,m}}{C_{V,m}} ,最終化簡為 \frac{dp}{p}+\gamma\frac{dV}{V}=0 ,解微分方程式可得
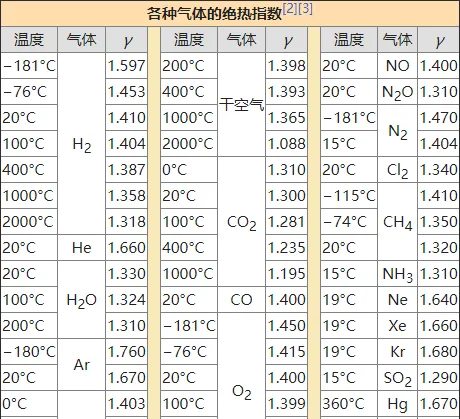
p_1V_{1}^{\gamma}=p_2V_{2}^{\gamma}=···=C (2-18)\gamma 稱為 比熱容比 或 絕熱指數 ,是一個聯系微觀與宏觀的系數。進一步我們有
\gamma=\frac{C_{p}}{C_{V}}=\frac{H}{U}>1 ; C_{p,m}=\frac{\gamma R}{\gamma-1} ; C_{V,m}=\frac{ R}{\gamma-1} (2-13.5)熱力學第二定律 (Second Law of Thermodynamics)的表述和推廣定理很多,也稱為熵增定律:
克耳文(Kelvin):不可能從單一熱源吸收熱量,使之完全變為有用功而不產生其他影響;克勞修斯(Clausius):不可能把熱量從低溫物體傳到高溫物體而不產生其他影響;
實質:在一切與熱相聯系的自然現象中它們自發地實作的過程都是不可逆的。
功能自發地、無條件地全部轉化為熱;但熱轉化為功是有條件的,而且轉化效率有所限制,也就是說功自發轉化為熱折一過程只能單向進行而不可逆。熱1說明了自然界中能量是守恒的(宏觀上),不會憑空產生,熱2則說明從單一熱源吸熱全部用來做功是不可能的,熱機效率不可能提高到100%,且存在其他理論極限,本文將在下節展開。

2.3卡農定理
熱力學第一、第二定律都是在一次次失敗制作永動機的嘗試中總結出來的。在真實的 熱力迴圈中(指系統或工作物質從初態出發經歷一系列的中間狀態最後回到原來狀態的過程 ),工作物質從高溫熱源吸熱所增加的內能不能全部轉化為對外做的有用功,它必須向外放出一部份熱。
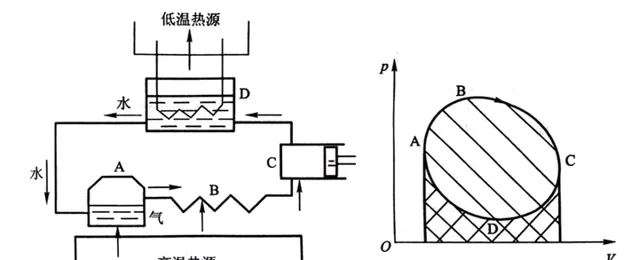
在上述準靜態熱機迴圈圖中,A→B過程溫度升高,內能增加,對外做功,屬於吸熱過程;B→C過程溫度降低,內能減少,對外做功,屬於放熱過程;C→D過程溫度降低,內能減少,外界對系統做功,屬於放熱過程;D→A過程溫度升高,內能增加,外界對系統做功,屬於吸熱過程。橫座標V軸正方向為對外做功,反方向系統對內做功;縱座標p軸正方向為吸熱,反方向為放熱。我們還可以發現:
1)熱機由熱驅動,熱比功多,因此是吸熱和升溫等價,放熱和降溫等價;
2)A→B→C過程的系統對外做功減去C→D→A過程的外界對系統做功,迴圈凈功就是p-V圖迴圈曲線圍成的面積;
3)順時針迴圈表示系統從高溫熱源吸熱,向低溫熱源放熱,系統對外界做凈功,此為熱機;
我們可以推出: 逆時針迴圈表示系統從低溫熱源吸熱,向高溫熱源放熱,外界對系統做凈功,此為冷機 。
我們用迴圈中 吸收的總熱Q1 及 放出的總熱Q2 計算熱機的理論效率
\eta_熱=\frac{Q_1-Q_2}{Q_1}=1-\frac{Q_2}{Q_1} (2-19)我們已經知道要使熱機迴圈,在經歷溫度升高、體積增加後必須要使溫度降低、體積減小才能回到初始狀態,而絕熱過程沒有系統與外界熱量交換,是一個理想的迴圈階段,因此在計算理論效率時必須有兩個絕熱過程(對外和對內做功)。根據現有理想瓦斯過程可以推匯出3種理想迴圈:卡農迴圈(等溫)、奧托迴圈(等體)、狄賽爾迴圈(等壓)。
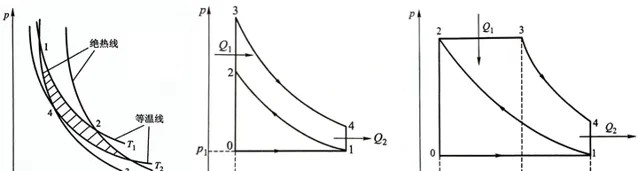
對於卡農迴圈:
1、恒溫可逆膨脹(1→2)有 Q_1=-W_1=nRT_1ln\frac{V_2}{V_1} , \Delta U_1=0 ;
2、絕熱可逆膨脹(2→3) W_2=\Delta U_2=nC_{V,m}(T_2-T_1) ;
3、恒溫可逆壓縮(3→4) Q_2=-W_3=nRT_2ln\frac{V_4}{V_3} , \Delta U_3=0 ;
4、絕熱可逆壓縮(4→1) W_4=\Delta U_4=nC_{V,m}(T_1-T_2) ;
整個過程系統對外界做功 -W=-(W_1+W_2+W_3+W_4)=nRT_1ln\frac{V_2}{V_1}+nRT_2ln\frac{V_4}{V_3} ,根據絕熱過程(2-18)的關系有 \frac{V_2}{V_3}=\frac{V_1}{V_4}=(\frac{T_2}{T_1})^\left( 1/(\gamma-1) \right) ,所以有 \frac{V_2}{V_1}=\frac{V_3}{V_4} ,所以 -W=nR(T_1-T_2)ln\frac{V_2}{V_1} ,根據熱機效率定義
\eta=\frac{-W}{Q_1}=\frac{nR(T_1-T_2)ln\frac{V_2}{V_1}}{nRT_1ln\frac{V_2}{V_1}}=1-\frac{T_2}{T_1} (2-20)結論有:
①卡農熱機熱效率僅與兩個熱源的溫度有關,溫差越高效率越大;
②因為-W=Q1+Q2,有 \frac{Q_1+Q_2}{Q_1}=\frac{T_1-T_2}{T_1} ,進一步有\frac{Q_1}{T_1}+\frac{Q_2}{T_2}=0 (2-21)
③ 卡農迴圈可逆,當迴圈逆向進行時η不變,外界對系統做功,把熱從低溫物體轉移到高溫物體 。
其余兩種迴圈在這裏僅做參考,效率都是和體積有關,用於內燃機更合適
奧托迴圈: \eta_{otto} =1-(\frac{V_1}{V_2})^\left(1-\gamma \right)
狄賽爾迴圈: \frac{V_1}{V_2}=K ,\frac{V_3}{V_2}=\rho , \eta_{diesel} =1-\frac{\rho^\gamma -1}{\gamma(\rho-1)K^{\gamma-1}}
卡農迴圈中兩個絕熱可逆過程的功相同,而兩個恒溫可逆過程的功不同,瓦斯恒溫可逆膨脹時因過程可逆是的熱機對外做功最大,而恒溫可逆壓縮時因過程可逆使系統從外界得功最小,故一個迴圈過程的總結果是熱機以極限的做功能力向外界提供了最大可用功,因而其效率時最大的。我們可以將其抽象為卡農定理:
(1)在相同的高溫熱源和相同的低溫熱源間工作的一切可逆熱機其效率相等,與工作物質無關;(2)在相同高溫熱源與相同低溫熱源間工作的一切熱機中,不可逆熱機效率不可能大於可逆熱機效率。
效率:100%>可逆>不可逆
在得到卡農定理後,我們開始建立一個新的狀態函式 熵S 。對於一個無限小的卡農迴圈,工質只從熱源吸收或放出熱đQ,結合式(2-21),
\frac{đQ_1}{T_1}+\frac{đQ_2}{T_2}=0
即任何卡農迴圈的可逆熱溫商之和為零。因過程量是可逆的,T是系統的函式恒不為0(熱3),在極限情況下有
\oint_{卡}^{ }\frac{đQ}{T}=0 (2-22)即任意可逆卡農迴圈的可逆熱溫商 \frac{đQ}{T} 沿封閉曲線的環路積分為0(沒有奇異點)。變量全微分的閉環路積分為零(保守場),反過來閉環路積分為零,所積變量為某函式的全微分。該變量的積分只取決於系統的始末狀態,因此為狀態函式
dS=đQ/T (2-23) 可逆時,對於 一般過程有dS≥đQ/T(克勞修斯不等式)結合熱1,đQ=dU+pdV可得
TdS=dU+pdV (2-24)因đQ是廣延量,T是強度量,因此熵S也是廣延量,但莫耳熵Sm時強度量。由於T>0,所以系統可逆吸熱đQ>0時,熵是增加的;系統可逆放熱,熵是減小的,單位[J/K]。為了計算熵,可用熱容表示
C_V=(\frac{đQ}{dT})_V=T(\frac{\partial S}{\partial T})_V ; C_p=(\frac{đQ}{dT})_p=T(\frac{\partial S}{\partial T})_p
對於理想瓦斯 dU=n C_{V,m} dT , 有dS= n C_{V,m} dT/T+nRdV/V ,進行T0到T的積分
S-S_0=nC_{V,m}ln\frac{T}{T_0}+nRln\frac{V}{V_0} (2-25)同理有
S-S_0=nC_{p,m}ln\frac{T}{T_0}-nRln\frac{p}{p_0} (2-26)熵增加原理:熱力學系統從一平衡態絕熱到達另一個平衡態的過程中,它的熵永不減少。
3、節流膨脹
理想模型在前面2章已經鋪墊的差不多了,接下來我們加快速度進入具體的原理階段。
3.1能量均分
之前推導比熱容比γ的時候,也把這個系數稱為絕熱指數,這個數不僅僅是推導過程中用來簡化方程式的書寫,還與很多真實瓦斯的分子類別型有關。對於單原子理想瓦斯,只有熱運動動能沒有勢能,由分子熱運動平均動能式(1-14) \bar{\varepsilon_{t}}=\frac{3}{2}kT ,以及理想瓦斯熱運動無擇優取向 \frac{1}{2}m\bar{v^{2}_x}=\frac{1}{2}m\bar{v^{2}_y}=\frac{1}{2}m\bar{v^{2}_z}=\frac{1}{3}\cdot\frac{1}{2}m\bar{v^{2}} 可知
\frac{1}{2}m\bar{v^{2}_x}=\frac{1}{2}m\bar{v^{2}_y}=\frac{1}{2}m\bar{v^{2}_z}=\frac{kT}{2} (3-1)單原子理想瓦斯的莫耳等體熱容為(1-15)和定體熱容定義
C_{V,m}=\frac{3}{2}R (3-2)上式說明在理想瓦斯中,xyz三個方向的平均平動動能都均分為kT/2。但是這一規律用來解釋雙原子及多原子瓦斯時,與實驗結果並不相符。
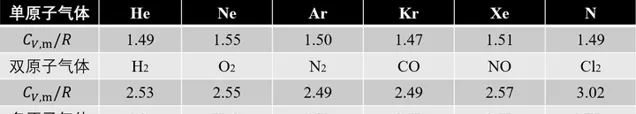
這是因為雙原子以上分子的分子結構不再能簡化成質點,必須把它們當成剛體。若要解釋單原子與多原子理想瓦斯熱容的差異,必須參照力學中自由度的概念。若一剛體在空間平動,又以各種可能轉動,因此需要3個平動、3個轉動自由度。對於雙原子分子,原子的品質集中在半徑 10^{-15} m的原子核上,而分子線度(化學鍵長度)為10^{-10} m,它繞自己化學鍵中心軸旋轉的轉動慣量僅為繞另外兩軸的10^{-10} 而可忽略,因此雙原子分子又3個 平動自由度 和2和 轉動自由度 。
若非剛性分子,相鄰兩原子間相對位置還可改變,原子間作用可使其振動,所對應的自由度為 振動自由度 。非剛性雙原子分子有一個沿兩質心連線振動的振動自由度,因此其自由度為6個。
N個原子組成的多原子分子自由度最多為3N個一般來說 ,在這3N個自由度中,有3個平動、3個轉動、3N-6個振動自由度。不過例外還是挺多的,振動一般都比較多,比如雙原子、直線三原子分子都是這樣。
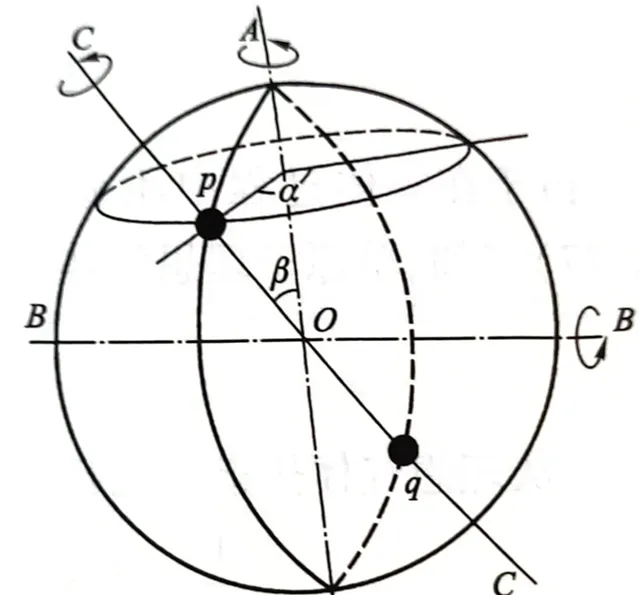
接下來把單原子理想瓦斯的平均平動動能推廣到多原子。單原子分子理想瓦斯的3個平均平動動能分到3個平動自由度中,每個自由度均分到kT/2平均動能,那麽我們也可以認為每一轉動及振動自由度也均分kT/2平均動能,有
處於溫度T的平衡態瓦斯中,分子熱運動動能平均分配到每一個分子的每一個自由度上的平均動能都是kT/2 。對於振動能量除動能外還有振動勢能,由於分子中原子所進行的振動都是振幅非常小的微振動,可以看作簡諧振動。一個周期內,簡諧振動的 平均動能 與 平均勢能 相等,所以 一個振動自由度均分kT的能量 。若某種分子有t個平動自由度、r個轉動自由度、v個振動自由度,則每一分子的總的平均能量為
\bar{\varepsilon}=(t+r+2v)\cdot\frac{1}{2}kT=\frac{1}{2}ikT (3-3)其中的 i=t+r+2v,表示的是分子的自由度 。需要強調的是,上式中的各種振動、轉動自由度都應是確實在對能量均分定理作全部貢獻的自由度,因為自由度會發生「凍結」。在常溫下 O_2 N_2 H_2 CO H_2O 等都是剛性分子,振動自由度被凍結,(只計算平動和轉動,雙原子=3+2,多原子=3+3)O_2 N_2 H_2 CO 的自由度i=5,水蒸氣H_2O 的自由度i=6。能量均分定理不僅適用於理想瓦斯,一般也可用於液體和固體,只不過凝聚態是透過分子間很強的交互作用來實作。
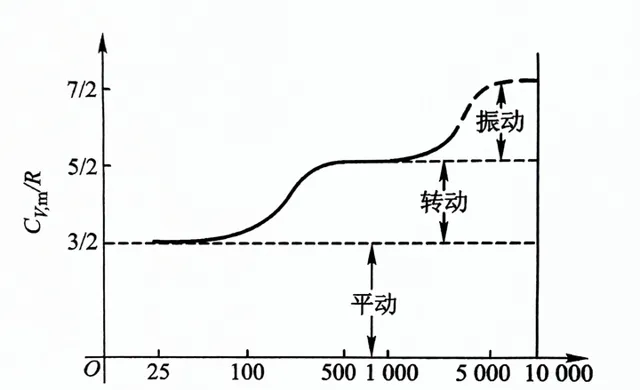
分子自由度和絕熱指數的關系有 γ=1+2/i ,比如常溫下不考慮振動,對於單原子i=3,γ=1.67;雙原子i=5,γ=1.4;多原子i=6,γ=1.33。
3.2傳熱方式
熱量傳遞有三種基本方式:熱傳導、熱對流與熱放射線。
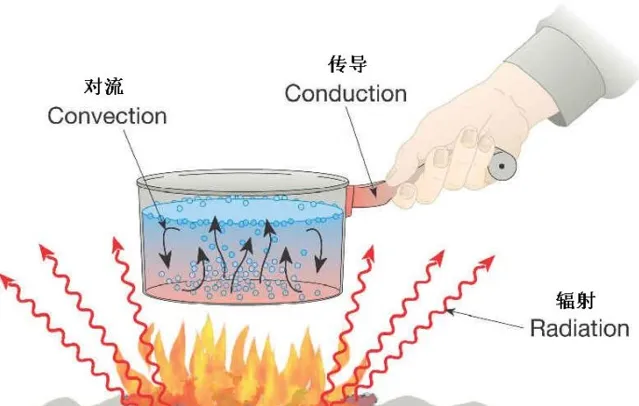
熱傳導 (Heat Conduction):是指在物體內部或相互接觸的物體表面之間,由於分子、原子及自由電子等微觀粒子的熱運動,而產生的熱量傳遞現象,是熱能 從高溫向低溫部份轉移的過程,是一個分子 向另一個分子傳遞振動能 的結果。主要透過傅立葉定律描述,主要有熱通量密度 {\display style {\overrightarrow {q}}} ,單位 W·m^{−2} , 熱導率k(熱導率為在單位時間內,每單位截面積所流過的熱量除以單位距離溫度變化量的負值) ,單位 W·m^{−1}·K^{−1} ,溫度梯度 {\display style {\big .}\nabla T{\big .}} ,單位K·m^{−1} 組成。
{\display style {\overrightarrow {q}}} =-k{\display style {\big .}\nabla T{\big .}} (3-4)熱對流 (Heat Convection):是指由於流體 的宏觀運動而引起的流體各部份之間發生相對位移(對流 ),冷熱流體相互摻混所引起的熱量 傳遞過程。對流傳熱可分為強迫對流和自然對流。強迫對流,是由於外界作用推動下產生的流體迴圈流動。自然對流是由於溫度不同密度 梯度變化,重力作用引起低溫高密度流體自上而下流動,高溫低密度流體自下而上流動。對流傳熱其經驗公式為牛頓冷卻定律,主要有流入或損失的熱量q,單位W,傳熱系數h(單位時間內透過單位面積傳遞的熱量),單位 W/(m^2K) ,傳熱面積A,單位m²,固體表面與周圍流體的溫度差△T,單位K。也可以忽略熱源面積將hA等效為α,稱為熱適應系數。
q=-hA\Delta T (3-5)熱放射線 (Thermal Radiation)是物體用電磁放射線 把熱能 向外散發的熱傳 方式,以熱放射線傳遞熱時 不需要介質 。任何物體溫度只要高於0K就會釋放熱放射線。熱放射線功率可以表示為
P_{rad}=\epsilon\sigma AT^4 (3-6)這裏只有兩個未知量,其中 斯特凡常數 可以用波茲曼常數表示,單位[ Wm^{-2}K^{-4} ],物體表面放射率 {\display style \epsilon } 介於0~1之間,黑體放射線為1。
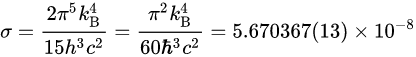
3.3焦耳-湯姆森效應
在制冷過程中,我們計算效率時要考慮的是從低溫熱源吸走的總熱量Q2,那麽外界必須對制冷機做功W,系統向高溫熱源放出的熱量為Q1,有 \eta_冷=\frac{Q_2}{W}=\frac{Q_2}{Q_2-Q_1} 。我們繼續逆時針卡農迴圈,同樣可得出
\eta_{卡冷}=\frac{T_2}{T_2-T_1} (3-7)可以看出制冷溫度越低時,制冷系數也越小,當T2為絕對零度時,則制冷系數為0。這裏補充一下
熱力學第三定律 (Third Law of Thermodynamics)又常被稱為 能斯特定理(Walther Hermann Nernst):
能斯特表述:系統的熵在等溫過程中的改變隨絕對溫度 趨於0。普朗克表述:在絕對 零度時,一切完美晶體的熵 值等於0。
推論:不可能透過有限的步驟使物體溫度降低到絕對零度。
1852年焦耳 James Prescott Joule 和湯姆森(克耳文勛爵, Kelvin Lord )發現瓦斯 會因在 等焓 的環境下膨脹,而使溫度上升或下降(基本上都是下降)。我們之前推導過等溫、等壓、等體、絕熱過程,我們這裏的絕熱都是可逆的,那也可以稱為等熵過程,現在還差一個等焓過程,這裏參考理想瓦斯的等某過程
等焓熱容C_H=\left( \frac{\delta Q}{\partial T} \right)_H
dH=dU+pdV+Vdp=\delta Q+Vdp
在等焓過程中 dH=\delta Q+Vdp=0
\left(\frac{\delta Q}{\partial T} \right)_H=-V\left( \frac{\partial p}{\partial T} \right)_H=-nR
C_H=-nR (3-8)等焓熱容是個負值,這意味著在等焓過程中溫度升高系統反而會對環境放熱 。
根據廣義帕松方程式(多方過程),等焓過程有
p_1V_{1}^{f}=p_2V_{2}^{f}=···=C (3-9)其中 f=2-1/γ,顯然可知f<γ ,至此所有等某過程介紹完畢。
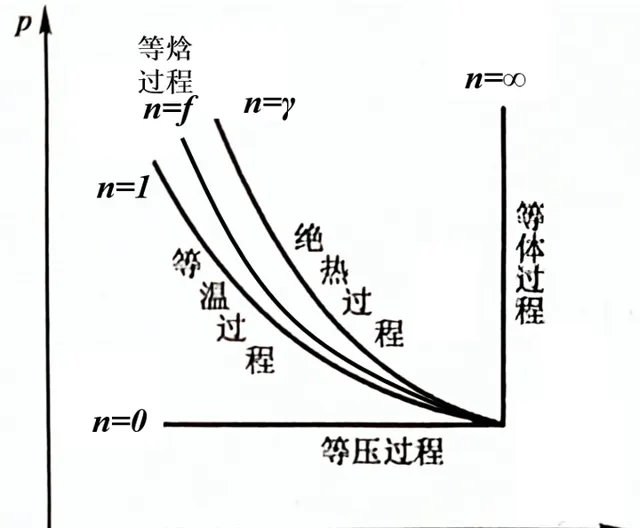
我們回頭看JT的實驗。如果系統隔熱效果好或者過程發生進行得很快,系統來不及和外界發生明顯的熱量交換,那我們也可以用絕熱過程近似分析。在這種絕熱條件下,高壓瓦斯經過多孔塞流到低壓一邊的穩定流動過程稱為 節流過程 。工業上常透過使瓦斯透過 節流閥 或 毛細管 來實作 節流膨脹 。

節流膨脹過程絕熱,所以Q=0。過程的功由兩部份組成,左側活塞運動至多孔塞的過程中,環境對系統做功 W_1=p_1V_1 ,右側活塞由多孔塞處移動至末態位置時,系統對環境做功 W_2=-p_2V_2 ,故整個節流膨脹過程功 W=p_1V_1-p_2V_2 ,根據熱1,有 U_1-U_2=p_1V_1-p_2V_2 ,整理得 U_1+p_1V_1=U_2+p_2V_2 ,即
H_2=H_1 (3-10)所以節流膨脹時等焓過程。對於真實瓦斯節流膨脹後不僅溫度會變化,壓力也要變化(不然無法自發進行),因此焓H(T,p)是T,p的函式,為描述瓦斯節流膨脹制冷或制熱能力的大小,引入如下焦耳-湯姆森系數,又稱 節流膨脹系數, 以下簡稱JT系數
\mu_{JT}=(\frac{\partial T}{\partial p})_H (3-11)我們對H(T,p)進行全微分 dH=(\frac{\partial H}{\partial T})_pdT+(\frac{\partial H}{\partial p})_Tdp=C_pdT+(\frac{\partial H}{\partial p})_Tdp , C_pdT 相表示理想瓦斯的焓變, (\frac{\partial H}{\partial p})_Tdp 表示瓦斯的非理想性帶來的附加貢獻,可以解釋為要克服分子間作用力而必須施加的功。
根據迴圈定理有 (\frac{\partial H}{\partial T})_p(\frac{\partial T}{\partial p})_H(\frac{\partial p}{\partial H})_T=-1 ,再重排變量有
(\frac{\partial H}{\partial p})_T=(\frac{\partial H}{\partial T})_p(\frac{\partial T}{\partial p})_H=-C_p\mu_{JT} (3-12) \mu_{JT} \mu_{JT}所以 dH=C_pdT-C_p\mu_{JT}dp
因為瓦斯膨脹dp<0,等焓過程dH=0,所以有下圖\mu_{JT} 為正時節流膨脹冷卻,反之則變暖。
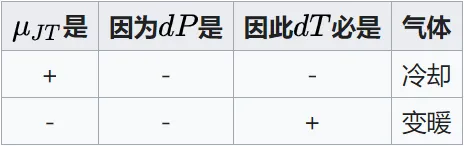
JT系數並不是一成不變的,對於真實瓦斯,隨著瓦斯壓力的升高,JT 系數由大變小,甚至出現負值。JT 系數從正值變到負值,必經過 JT 系數等於零的點。當 JT 系數為正值,即 \mu_{JT} >0,發生致冷效應;當 JT 系數為負值,即\mu_{JT} <0,發生致熱效應;當 JT 系數為零,即\mu_{JT} =0,為理想瓦斯,既不致冷,也不致熱。可透過\mu_{JT} =0的點來判斷\mu_{JT} 的正負,將一系列\mu_{JT} =0 的點連線起來就形成一條曲線,稱為焦耳-湯姆遜反轉曲線(JTIC),即透過此曲線時,焦耳-湯姆遜效應反轉。透過 JTIC 很容易判斷出實際瓦斯的致冷區域和致熱區域。在低壓側,即在 JTIC 內側是致冷區域;在高壓側,即 JTIC 外側是致熱區域。
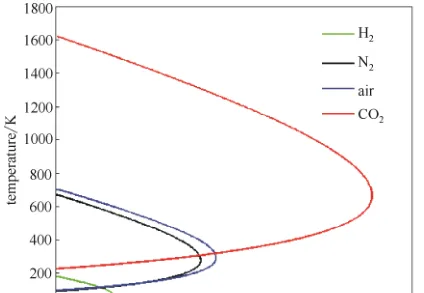
可以這樣理解,在低壓側,當瓦斯膨脹,分子 之間的平均距離上升。因為分子間吸重力,瓦斯的位能 上升。因為這是等焓過程,系統的總能量守恒 ,所以位能上升必然會令動能 下降,故此溫度下降;高壓側,當分子碰撞 ,動能暫時轉成位能。由於分子之間的平均距離上升,每段時間的平均碰撞次數下降,位能下降,因此動能上升,溫度上升。在制冷過程中我們需要的當然是低壓側的效應。
4、制冷劑迴圈
我們已經解鎖了全部的物理化學過程前置,實作了從-1到0的改變,接下來我們正式開始介紹空調。
4.1四大裝置
實作空調變冷迴圈的制冷劑五花八門,無論是壓縮式還是吸收式,不管是非等溫流還是解不出的復雜亂流,都必須遵循熱力學第二定律,參照之前的卡農熱機迴圈,我們在這裏給出卡農冷機迴圈:
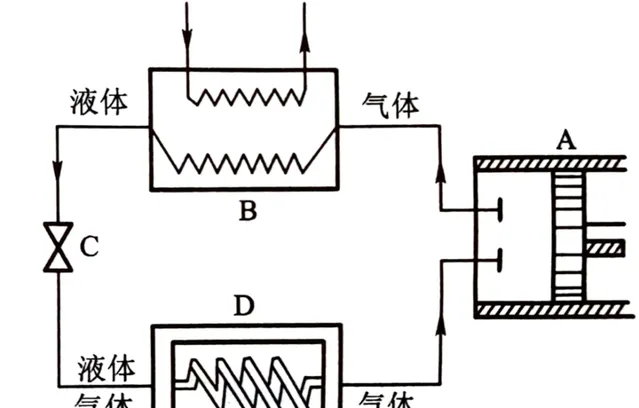
1)氣態制冷工質被壓縮機A壓縮後成為 高溫高壓蒸汽 ;
2)壓縮蒸汽進入冷凝器B中冷卻後成為 中溫高壓 後釋放汽化熱(相變焓)成為 液體 ,釋放Q1熱量;
3)中溫高壓液體進入節流裝置C進行等焓節流膨脹成為 低溫低壓濕蒸汽(氣液混合態) ;
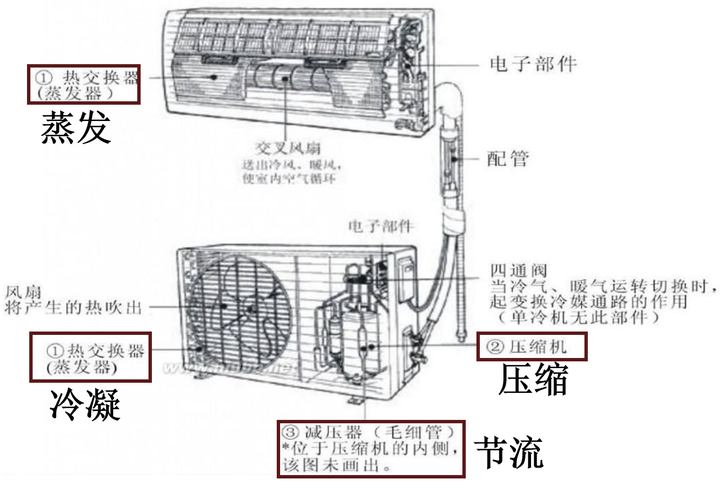
4)低溫低壓濕蒸汽進入蒸發器D中,吸收Q2熱量,全部蒸發為 低溫低壓過熱蒸汽 (仍比蒸發風低),隨後進入下一迴圈。
這裏由於外部做功,制冷效率算出來會大於1,因此這個值也稱為制冷系數,制冷劑經過的ABCD四個裝置是市面上絕大多數空調所必須具備的。其中壓縮機A現在已經都是密閉式渦旋壓縮機;冷凝器B其實就是一排排線圈纏繞管,配合冷凝風扇向室外散熱;冷凝管出來後的液體去節流裝置C前還有經歷一遍幹燥過濾,節流閥多為熱力膨脹閥或電子膨脹閥;節流膨脹後的低溫低壓濕蒸汽經過蒸發器D,和冷凝器相同的線圈和風扇,只不過是向室內吹風,重新變回低溫低壓過熱蒸汽。
ABC一般放空調外機,D一般放在空調內機。
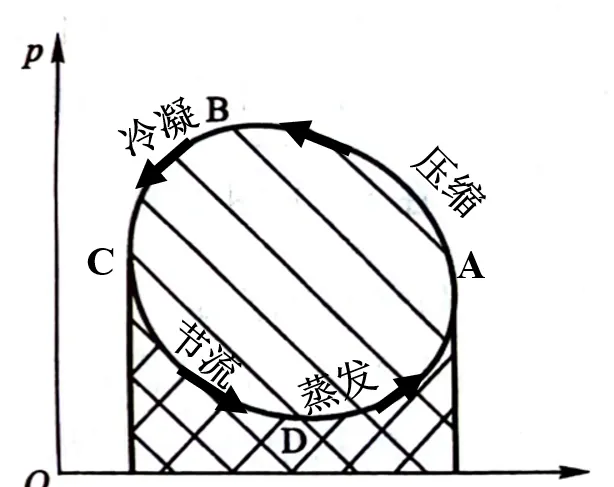
為了更加準確計算真實制冷劑的效率,需要借助壓焓圖判斷工質在各個機組的狀態,這裏僅做定性介紹。
壓焓圖曲線的含義可以用一點(臨界點)、二線(飽和液體線、飽和蒸汽線)、三區(液相區、兩相區、氣相區)、五態(過冷液狀態、飽和液狀態、過熱蒸汽狀態、飽和蒸汽狀態、濕蒸汽狀態)和八線(等壓線、等焓線、飽和液線、飽和蒸汽線、等幹度線、等熵線、等比體積線、等溫線)來概括。
這裏可以參考空調壓焓圖
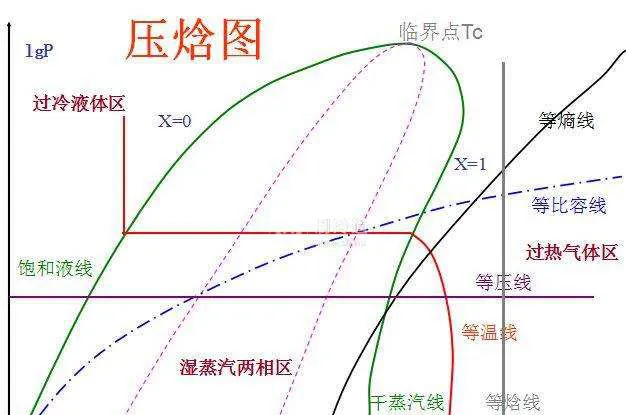
很抽象對不對,其實並不需要記憶多少概念,我們結合實際的迴圈來看,以R407C制冷劑為例。在單級壓縮焓圖中:A點對應的是11℃的過熱蒸汽(焓h=412kJ/kg);壓縮至B點55℃的高溫高壓蒸汽(焓h=432kJ/kg);冷凝器冷卻後為中溫高壓液體,36℃達到飽和(焓h=245kJ/kg)C點31攝氏度達到過冷(焓h=238kJ/kg);節流膨脹降壓過程等焓,D點溫度降低至6℃的低溫低壓濕蒸汽;蒸發器蒸發後重新變成11℃的過熱蒸汽(焓h=412kJ/kg)。

壓縮焓圖中,蒸發後的飽和蒸汽並不是達到飽和蒸汽線(幹蒸汽線)就開始壓縮,這個過熱蒸汽是防止壓縮機被液化擊穿,一般為5℃;脹閥後,必須要在蒸發器內將液體變為瓦斯,因此在節流膨脹前要進行過冷卻過冷度一般為5℃。
過熱度:吸入瓦斯溫度-蒸發溫度過冷度:冷凝溫度-膨脹閥前溫度
4.2冷熱模式
我們知道功可以自發轉變為熱量,但是這種轉變太慢。我們把卡農冷機反過來,交換一下制冷劑的迴圈順序,使壓縮機把壓縮後的高溫高壓瓦斯進入蒸發器,蒸發器本來就是向室內吹氣,這樣把高溫制冷劑熱量透過強迫熱對流傳遞給室內,似乎2個裝置就可以完成迴圈。但是,還記卡農定理嗎?這個溫差簡直就是能耗黑洞。而就現實來說,沒有節流降壓過程,蒸發散熱後的液體更不可能被壓縮,所以必須經過完整的卡農迴圈。
我們按照相反過程推導:
1)氣態制冷工質被壓縮機A壓縮後成為 高溫高壓蒸汽 ;
2)壓縮蒸汽進入蒸發器D中冷卻後成為 中溫高壓 後釋放汽化熱(相變焓)成為 液體 ,釋放Q1熱量;
3)中溫高壓液體進入節流裝置C進行等焓節流膨脹成為 低溫低壓濕蒸汽(氣液混合態) ;
4)低溫低壓濕蒸汽進入冷凝器B中,吸收Q2熱量,全部蒸發為 低溫低壓過熱蒸汽 (仍比蒸發風低),隨後進入下一迴圈。
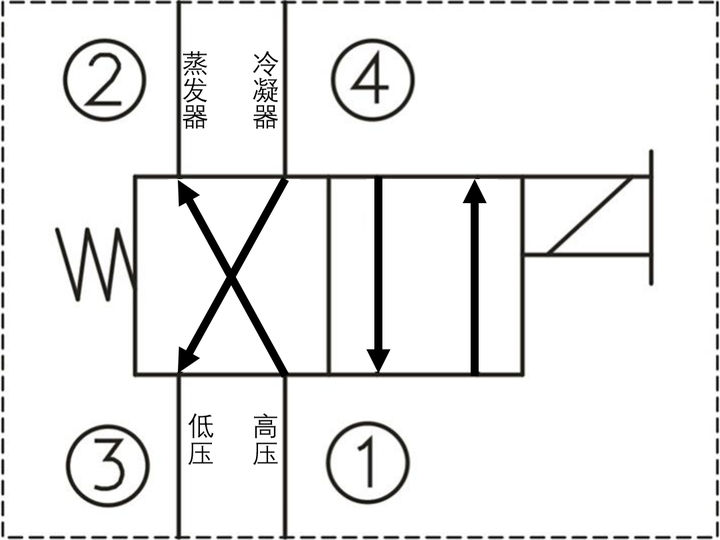
這實質上也是 從低溫熱源吸熱,向高溫熱源放熱 的冷機過程。為了實作冷熱模式交替,需要切換制冷劑流經蒸發器和冷凝器的方向,這裏我們可以直接使用一個二位四通閥。對於壓縮機,我們不能弄錯高壓和低壓的流向,必須時制冷劑總是從低壓進高壓出,同樣對於膨脹閥和幹燥器,我們也要設定一個二位四通閥,使得總是節流膨脹,這樣一來,我們空調開啟制冷模式時四通閥是一個位,開啟制熱模式時四通閥又是一個位。不過對於很多地方,空調變熱時目標溫差要遠大於制冷時的目標溫差,所以在具有制熱模式的空調蒸發器風扇外增設了電熱管,進一步改善制熱效果。
後面的溫度調節,電控方法之類的我覺得不是原理,更偏套用,就不展開了。
5、番外篇
基於節流膨脹的制冷已經被開發一百多年了,盡管這些年也誕生了很多非主流制冷方式,但空調依舊是比別的制冷方式節能。
5.1空氣液化
在空調變冷劑迴圈中,我們都是用沸點比較高(室溫能達到)的制冷劑作為例子,它們在經過冷凝器後就已經是液態,節流膨脹只是讓其溫度進一步降低,試想一下,我們如果我們不要蒸發過程了,只要控制溫壓在JT反轉曲線內,是可以繼續降溫的,但是如果需要使用節流效應進行 等焓液化 ,我們需要看另一個圖——溫熵圖。
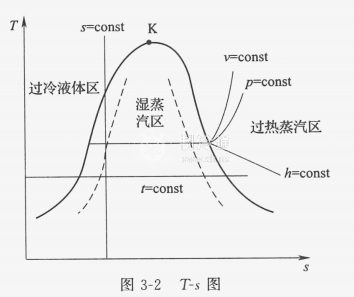
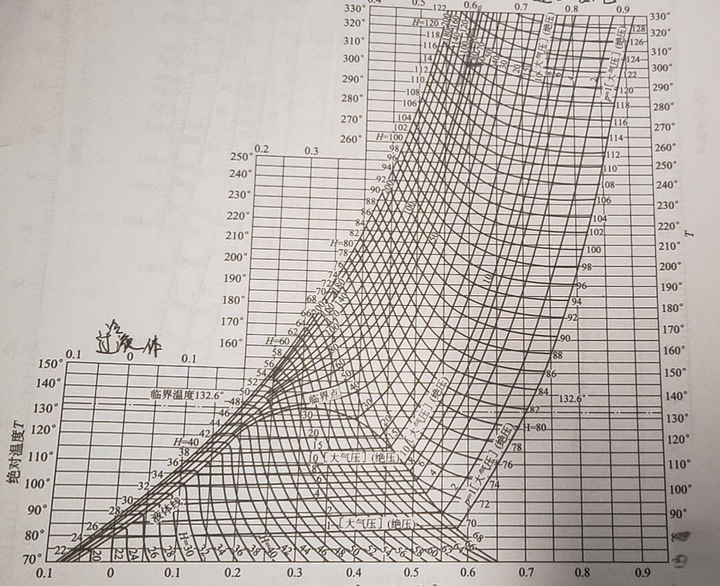
溫熵圖和之前提到的壓焓圖有點像,也分為過冷液體區、濕蒸汽區和過熱蒸汽區,也有臨界點,而且實際上用途更廣泛。首先是 等溫線 和 等熵線 ,它們就是和橫縱座標平行的線,如果用溫熵圖描述卡農熱機,我們就可以得到一個標準的矩形。 等壓線 :在濕蒸汽區近乎與等溫線重合;在過熱蒸汽區為 左下至右上曲線 ;過冷液體區,等壓線密集於飽和液體線附近,可近似以飽和液體線代替。 等焓線 :為 左上至右下曲線 ,濕蒸汽區坡度大,過熱蒸汽區較平坦。此外還有等溶線、等幹度線等。上面提到的液化實際上就是氣態經等焓線進入氣液兩相區的過程。比如下面的空氣溫熵圖中,我們在10MPa(100個大氣壓)下時,溫度要低於169.4K才能沿等焓線液化。
目前液化空氣的方法有林德(Linde)過程和克勞德(Claude)過程。林德過程也就是林德-漢普遜迴圈。原理如下,①透過壓縮加熱瓦斯,以給予其參與迴圈所需的外部能量。②將瓦斯浸入低溫環境的方式將其冷卻,使其失去一部份熱量;③節流膨脹,現階段瓦斯達到整個過程的最低溫度,將再度迴圈並被送回;④送回後的瓦斯經換熱器冷卻下一階段的返流瓦斯;⑤回到階段①開始下一迴圈。在這個簡圖中,似乎只有外界對系統做功,溫度應該是上升,實際上冷凝是有另一套迴圈的,我們可以在溫熵圖中進一步深入。
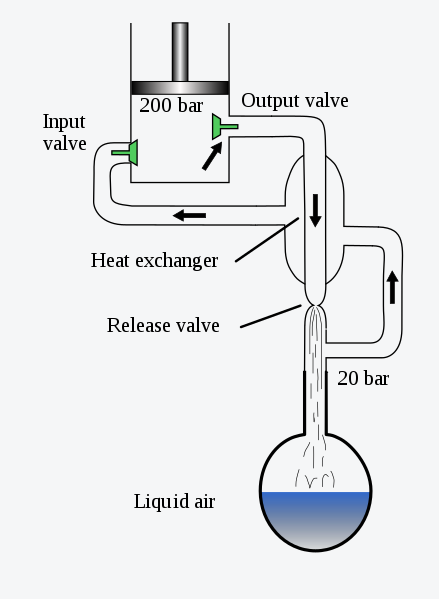
在林德迴圈中: 狀態1 處於壓力為p1的瓦斯,經壓縮機壓縮至壓力p2後等壓冷卻至 狀態2 ,該瓦斯經逆流換熱器被分離返回後的瓦斯進一步等壓冷卻至 狀態3 ,再經節流效應等焓液化至濕蒸汽區的 狀態4 ,在貯分離器中液化後 狀態0 的液態會被分離器不斷匯出,未被液化的 狀態5 瓦斯被匯出經過逆流換熱器給下一波狀態2瓦斯冷卻後再和狀態1的瓦斯一起被壓縮。這個過程冷卻和換熱的液體都是隨著迴圈進行溫度越來越低的,因此可以將空氣液化。
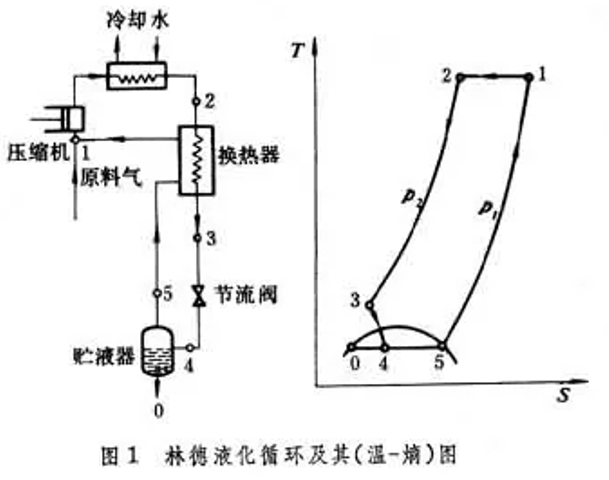
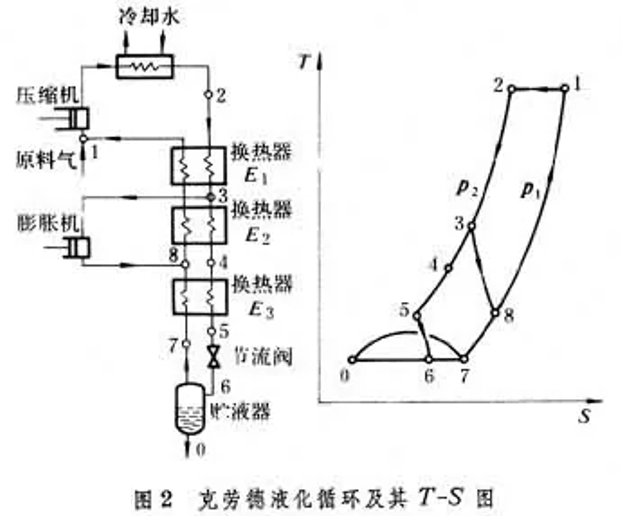
節流膨脹不對外做功,而絕熱膨脹可對外做功,因此理論上可制取更低溫度,但低溫液化會造成損失,因此膨脹機必須與節流閥共同使用,為此克勞德迴圈增加了2個換熱器和1個膨脹機。我們從狀態3開始分析,其中的一部份瓦斯沿3→8絕熱膨脹至 狀態8 ,另一部份經換熱器2(狀態4)、換熱器3(狀態5)、節流閥(狀態6)後,液化後的狀態0,未液化的狀態7都與林德迴圈相同,狀態7瓦斯經逆流換熱器3後與膨脹機出口狀態8匯合,匯合後的瓦斯經換熱器1、2後進入壓縮機進入下一個迴圈。
5.2半導體制冷
之前所有的降溫過程都或多或少用上了JT效應或者膨脹做功,那麽接下來我們看一點不一樣的。
熱電效應( 英語:Thermoelectric effect)是一個由溫差產生電壓的直接轉換,且反之亦然。簡單的放置一個熱電裝置,當他們的兩端有溫差時會產生一個電壓,而當一個電壓施加於其上,他也會產生一個溫差。
1821年塞貝克發現將二種不同金屬 各自的二端分別連線構成的回路,如果兩種金屬的兩個結點處溫度不同,就會在這樣的路線內發生電流。這種現象稱為 賽貝克效應 (Seebeck Effect)。
1834年珀爾帖 發現了與塞貝克效應 的相反效應,即當電流流經兩個不同導體形成的接點時,接點處會產生放熱和吸熱現象,放熱或吸熱大小由電流的大小來決定。這就是 珀爾帖效應 (Peltier Effect)。
1854年威廉·湯姆森 將一根導線通恒定電流,由於導線有電阻而發熱。再將這根帶電的導線的某小局部加熱;使它產生溫度梯度 。這根導線就在原有發熱的基礎上,出現吸熱或放熱的現象。或者反過來,當一根金屬棒的兩端溫度不同時,金屬棒兩端會形成電勢差。這就是 湯姆森效應( Thomson effect)
在只有金屬導電的年代,這幾個效應確實只能叫「熱」電效應,但是在半導體時代,我們有更好的用途。根據塞貝克效應我們可以得到塞貝克系數的定義:當對材料施加溫度梯度時,以及當材料達到電流密度 處處為零的穩定狀態時,建立的電壓。
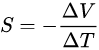
從物理上講,塞貝克系數的大小和符號可以理解為材料中電流攜帶的每單位電荷的熵。它可能是正的或負的。對導體中獨立移動的自由電荷載流子 ,塞貝克系數對於帶負電的載流子(例如電子 )為負,對於帶正電的載流子(例如電子電洞 )為正。
塞貝克效應通常主要由電荷載流子擴散的貢獻決定,電荷載流子擴散往往會將電荷載流子推向材料的冷側,直到建立補償電壓。因此,在p型半導體 , S 為正,在n型半導體 (僅具有負移動電荷,電子 )中, S 為負。然而,在大多數導體中,電荷載流子表現出類電洞和類電子行為,並且 S 的符號通常取決於它們中的哪一個占主導地位。
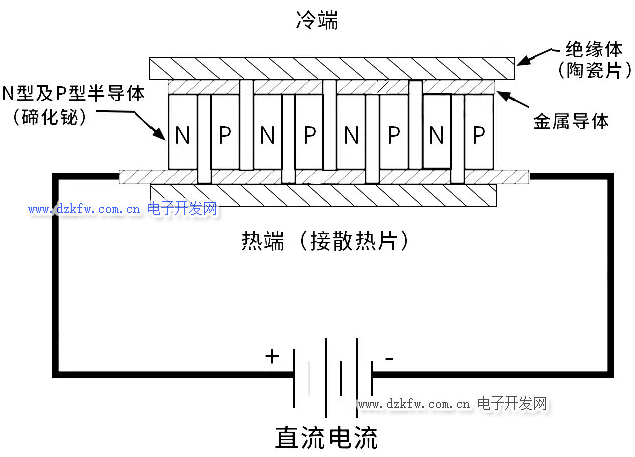
對於一般金屬導體和N型半導體,都是電勢高的地方為熱端,電勢低的地方為冷端,而P型半導體則相反。根據這個效應,我們將很多個這樣的半導體聯結成電偶對時,在這個電路中接通直流電流後,就能產生能量的轉移,電流由N型元件流向P型元件的接頭吸收熱量,成為冷端;由P型元件流向N型元件的接頭釋放熱量,成為熱端。
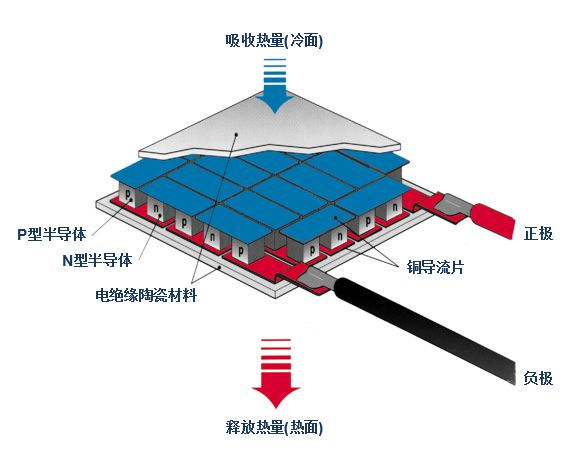
這樣的元器件就是半導體制冷器(Thermoelectric cooler),不過若要使用它來制冷,得準備比它自身大得多的風扇,而且這個只能用於熱傳導接觸制冷,因為實在是帶不起風扇吹出的制冷量。

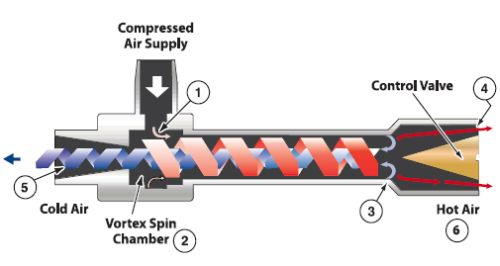
5.3渦流管制冷
這裏參考渦流管到底怎麽制冷的
渦流管制冷是一種借助渦流管的作用使高速氣流產生漩渦分離出冷、熱兩股氣流,利用冷氣流而獲得制冷方法。渦流管其實和上面的TEC很像,就是一端制冷另一端制熱,只不過對於民用制冷來說使用條件更加嚴格,需要3-10bar的進氣壓力,通常為7bar,這導致沒有壓縮空氣的地方必須要帶一個空壓機,噪音就太大了。
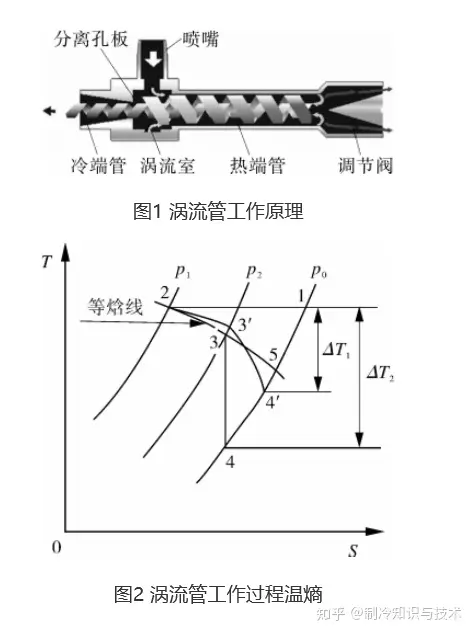
我們再來看能耗,也就是渦流管的溫熵圖,圍成面積對應因次為能量。
圖中p0、p1、p2分別為環境大氣壓、噴嘴出口壓力、空壓機出口壓力,ΔT1為實際溫降,ΔT2為理論最大溫降。1-2-3-4-1為理想渦流管的制冷迴圈,其面積為理想渦流管制冷量,環境空氣進入空氣壓縮機等溫壓縮,後經噴嘴的節流過程,進入渦流管絕熱膨脹,最終排出冷熱端管。
圖中2-3'為實際噴嘴節流降溫過程,過程中摩擦等損失使焓值增加,3'-4'為實際渦流室中瓦斯膨脹降溫過程,其膨脹效率介於絕熱膨脹與絕熱放氣之間,焓減小一部份轉化為瓦斯的推動功,因而其制冷效率總是比理想節流的等焓過程高。1-2-3'-4'-1為實際渦流管制冷迴圈,其面積為實際渦流管制冷量。2-5為等焓線,1-2-5過程為單一利用空氣壓縮機出口瓦斯進行節流制冷迴圈的制冷量。從圖可以發現理想及實際渦流管總的制冷量總是比單一運用節流效應制冷的制冷高。
但是,實際上空調並不需要渦流管如此猛烈的溫降,而且渦流管不像空調把空氣迴圈和制冷工質迴圈分開,這導致在空氣凈化上還是得額外走一路,高溫瓦斯回收困難,綜合成本下來還是空調最劃算。