
摘要
目的: PCSK9主要由肝脏分泌到循环中,与低密度脂蛋白受体(LDLR)的同源和非同源受体(包括CD36)相互作用,从而有利于它们在细胞内降解。由于PCSK9缺乏会增加脂质和脂蛋白受体的表达,从而促进细胞脂质积累,我们研究了这是否会影响心脏代谢和功能。
方法和结果: 用标准脂肪饮食喂养野生型(WT)、Pcsk9 KO、肝脏条件性Pcsk9 KO和Pcsk9/Ldlr双KO雄性小鼠20周,然后评估运动耐量、肌肉力量和心脏特性。 Pcsk9 KO表现为跑步耐量减低,并伴有超声心动图异常,提示射血分数保留的心力衰竭(HFpEF)。 与WT小鼠相比,在最大耦合和非耦合呼吸后,Pcsk9KO小鼠的心脏线粒体活性降低,并且与心脏代谢的主要变化以及LDLR和CD36的表达增加以及脂质积累有关。在Pcsk9/Ldlr DKO中观察到类似的表型,因此排除了LDLR导致Pcsk9 KO小鼠中观察到的心脏损伤的可能。肝脏选择性Pcsk9 KO模型的心脏功能分析进一步排除了循环PCSK9在HFpEF发展中的作用,表明局部产生的PCSK9可能发挥作用。同时,与匹配的对照受试者相比,PCSK9的R46L功能丧失变体的携带者左心室质量增加,但射血分数相似。
结论: PCSK9缺乏通过一种不依赖LDLR的方式影响心脏脂质代谢,并有助于HFpEF的发展。
名词解释
PCsk9(proprotein convertase subtilisin / kexin type 9): 前蛋白转化酶枯草溶菌素9
HFpEF(heart failure with preserved é jection fraction): 射血分数保留的心力衰竭。射血分数保留的心衰(HFpEF)指心脏射血分数正常或接近正常(>0.5或0.45)但有症状或体征和临床表现的心力衰竭,其并非一种特异性的诊断或综合征,它是由多种疾病(排除非源性因素)引起的一组症候群。
ATP(adenosine triphosphate): 三磷酸腺苷
LDLR(low - density lipoprotein receptor): 低密度脂蛋白受体
VLDLR(very low - density lipoprotein receptor): 极低密度脂蛋白受体
WT(wild - type): 野生型
FA(fatty acid): 脂肪酸
LPL(lipoprotein lipase): 脂蛋白脂肪酶
PPARα(peroxisome proliferator-activated receptor alpha): 过氧化物酶体增殖剂激活受体α
PPARγ(peroxisome proliferator-activated receptor gamma): 过氧化物酶体增殖剂激活受体γ
TAG(triacylglycerol): 甘油三酯
GPI( glycosylphosphatidylinositol): 糖基磷脂酰肌醇
LVPW(left ventricular posterior wall): 左心室后壁
ETC(electron transport chain): 电子传递链
前言
心脏主要利用有氧代谢来满足其能量需求,其中大部分三磷酸腺苷(ATP)是在脂肪酸(FA)氧化后产生的。
与肝脏不同,心脏不能合成大量FA,因此,FA的需求主要从循环中摄取来支持,其中FA以游离FA形式与白蛋白结合或以脂蛋白中的甘油三酯形式运输。然后通过脂蛋白脂肪酶(LPL)的活性将FA从白蛋白中解离或从脂蛋白中释放出来,并通过FA转运蛋白或FA转位酶(如CD36)递送至心肌细胞。脂蛋白通过与包括极低密度脂蛋白受体(VLDLR)在内的细胞表面特定受体相互作用,将FA和胆固醇输送至心肌细胞。
心脏脂质需求是一个精细调节的过程,它平衡脂质摄取和线粒体β-氧化以支持心脏代谢,同时需要防止过度的脂质累积——反而会导致心肌细胞功能障碍。在心力衰竭患者中,心肌细胞转为糖酵解作为ATP生成的首选途径,从而促进细胞三酰甘油(TAG)的累积。
在代谢综合征和糖尿病患者中,乳酸、酮体或氨基酸被用作替代FAs的能量来源。在分子水平上,一方面TAG累积和FA超载促进心肌细胞中的线粒体功能障碍和氧化磷酸化解偶联,另一方面促进脂毒性物质的产生,包括二酰基甘油、长链酰基辅酶A、酰基肉碱和溶血磷脂,导致心力衰竭。
一些实验观察提供了脂质累积和心脏功能障碍之间的联系。缺乏脂肪甘油三酯脂肪酶的小鼠表现出明显的心脏TAG累积,同时伴有心力衰竭和过早死亡。相似地,过氧化物酶体增殖物激活受体α(PPARa)和γ(PPARγ)的过度表达会增加FA氧化,但导致FA摄取超过氧化引起的心脏脂质代谢失衡,从而导致脂质累积。
与之一致的,在心肌细胞中选择性表达糖基磷脂酰肌醇(GPI)锚定的人LPL(aMHC-LpLGPl)小鼠增加了心脏摄取和来自循环脂蛋白(包括甘油三酯、FA和胆固醇)的脂质累积。
所有这些数据都与以下概念一致,即促进参与脂质摄取途径的活性可能有助于心脏脂质超负荷和毒性,并表明增加心脏中脂蛋白受体表达的因素可能导致心脏功能障碍,如缺氧诱导VLDLR介导的心脏脂毒性。
前蛋白转化酶枯草杆菌蛋白酶/kexin 9型(PCSK9),一种692个氨基酸的糖蛋白,已知可控制低密度脂蛋白受体(LDLR)再循环。除LDLR外,PCSK9还靶向LDLR的同源和非同源受体,包括VLDLR15、ApoER2(LRP8)、LRP1和CD36,并通过抑制内体﹣溶酶体区室中脂蛋白的解离来破坏它们的再循环,从而促进他们的退化。因此,增加的PCSK9水平会导致这些受体的再循环减少,这一发现在肝脏中得到了广泛的研究。
然而, PCSK9可发挥肝外效应:它的缺乏与胰腺细胞功能受损和人类患糖尿病的风险增加相关。 值得注意的是,Pcsk9 KO小鼠的体重增加和内脏脂肪组织沉积,这一发现在PCSK9功能丧失变体的携带者中得到证实。观察到这些受试者还表现出心外膜脂肪组织增加,再加上PCSK9在控制参与心脏脂蛋白摄取的关键受体中的关键作用,为研究PCSK9缺乏是否影响心脏脂质代谢和功能提供了理论依据。
为此,我们在保留胰岛素反应的实验条件下测试了PCSK9缺乏对心脏代谢和功能的影响。以标准脂肪饮食喂养Pcsk9 KO小鼠20周,我们观察到PCSK9缺乏会影响心脏中的脂质代谢和能量产生,导致左心室壁增厚和射血分数保留的心力衰竭(HFpEF)的进展。为了进一步研究这一观察结果在临床环境中的影响,在携带PCSK9功能丧失多态性的受试者中进行了心功能研究。
方法
关于小鼠、超声心动图分析、疲劳测试、前肢抓握测试、耗氧率、代谢组学、蛋白质组学、蛋白质组学、蛋白质印迹分析、心脏组织中胆固醇积累的分析、血浆剂量、生物信息学和统计分析的详细描述于在线补充材料中提供。
结果
PCSK9缺乏与射血分数保留的心力衰竭有关
为了研究PCSK9对全身和细胞脂质和脂蛋白代谢的影响是否可能影响心脏功能,我们评估了喂食标准脂肪饮食20周的PCSK9 KO和野生型(WT)雄性小鼠的心脏形态。Pcsk9 KO小鼠心脏的心超分析(图1A)显示,与WT小鼠(图1B和C)相比,在收缩期和舒张期期间左心室后壁(LVPW)的厚度增加,尽管两组之间的总体重没有差异(在线补充材料,图S1A)。
尽管与野生型小鼠相比,Pcsk9 KO小鼠中的左心室质量(图1E)和射血分数(图1F)的归一化相似,但相对壁厚增加(图1D),因此提示心脏存在同心重构。为了评估这种情况是否与心脏功能受损有关,在WT和Pcsk9 KO 小鼠中行疲劳测试中的运动不耐受和跑步耐量试验。与WT小鼠相比,后者在疲劳测试中观察到的跑步距离和跑步时间显著减少(图1G和H)。
值得注意的是,这种表型不是骨骼肌性能降低的结果,因为在前肢抓握测试(图1I)中观察到了类似的结果,并且Pcsk9 KO和WT小鼠之间比目鱼肌的耗氧率没有差异(在线补充材料,图S1B)。这些数据表明Pcsk9 KO呈现出HFpEF的特征。
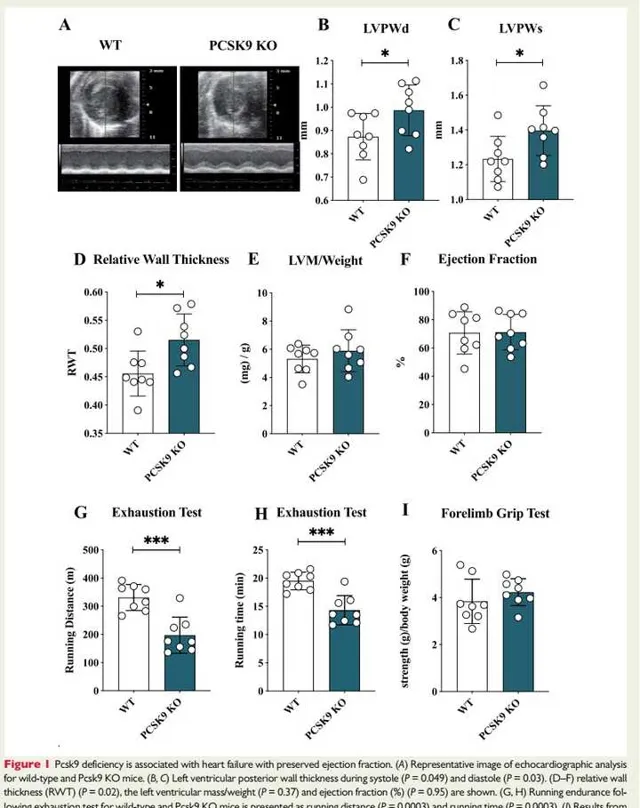
图1:Pcsk9缺乏与射血分数保留的心力衰竭有关。
图1A:WT和Pcsk9 KO小鼠超声心动图分析图像。
图1B和C:分别为收缩期和舒张期期间的小鼠左心室后壁厚度(LVPW)。结果发现:与WT小鼠相比,在收缩期和舒张期期间Pcsk9 KO小鼠LVPW的厚度增加。
图1D:为心超显示的相对壁厚,结果提示:与WT小鼠相比,Pcsk9 KO小鼠相比壁厚增加。
图1E:为左心室质量/体重,提示与WT小鼠相比,Pcsk9 KO小鼠该比值增大(尽管未达到统计学差异)。
图1F:为射血分数(%),结果提示,Pcsk9 KO小鼠与WT小鼠的射血分数无显著差异。
图1G和H:为WT和Pcsk9 KO小鼠在疲劳测试的跑步耐力,表现为跑步距离(1G)和跑步时间(1H)。结果提示:与WT小鼠相比,Pcsk9 KO小鼠在疲劳测试中观察到的跑步距离和跑步时间均显著减少。
图1:显示了前肢抓地力测试的结果,提示WT和Pcsk9 KO小鼠骨骼肌性能近似。综上,Pcsk9 KO小鼠呈现射血分数保留的心力衰竭,提示Pcsk9缺乏与射血分数保留的心力衰竭有关。
PCSK9缺陷小鼠的心脏线粒体代谢发生改变
鉴于PCSK9在细胞脂质生物学中的关键作用,我们接下来评估PCSK9缺乏对心脏脂质代谢的影响及其与心脏能量需求的相关性。与最大耦合和非耦合呼吸下的对照小鼠相比,来自Pcsk9 KO小鼠的新鲜分离的心脏表现出氧消耗率的显著降低(图2A)。这一发现,加上在Pcsk9 KO小鼠中观察到的电子传递链(ETC)复合物11的活性降低(在线补充材料,图S2A和B),促使我们测试 ATP 的产生是否受到影响。
与WT同窝对照以及其他参与能量产生的辅助因子相比,Pcsk9 KO 鼠心脏中的ATP水平(图2B)和ATP能量电荷(图2C)降低(在线补充材料,图S2D-K)。此外心脏组织的详细蛋白质组学分析表明,与WT(在线补充材料,图S2A)相比,Pcsk9 KO心脏中几种线粒体蛋白的表达受到影响,包括ETC复合物的关键结构成分(图2D)。
通过对代表每个线粒体复合物的蛋白质(包括NADH泛醌氧化还原酶亚基B8(Ndufb8﹣复合物Ⅰ)、琥珀酸脱氢酶复合物亚基B(Sdhb-复合物Ⅱ)和泛醇-细胞色素C还原酶核心蛋白2(Uqcrc2-complex Ⅲ))进行蛋白质印迹分析,发现与WT同窝对照相比,在Pcsk9 KO小鼠的心脏中上述蛋白表达显著减少(图2E和F),从而进一步证实了这一发现。
这一发现连同复合物Ⅱ活性的降低(在线补充材料,图S2B)支持FADp进入ETC效率较低这一结果的可能性,该过程可能会影响FA分解代谢。同时,线粒体DNA拷贝数也减少(在线补充材料,图S2C),表明Pcsk9 KO小鼠的心脏中线粒体功能和含量受到影响。
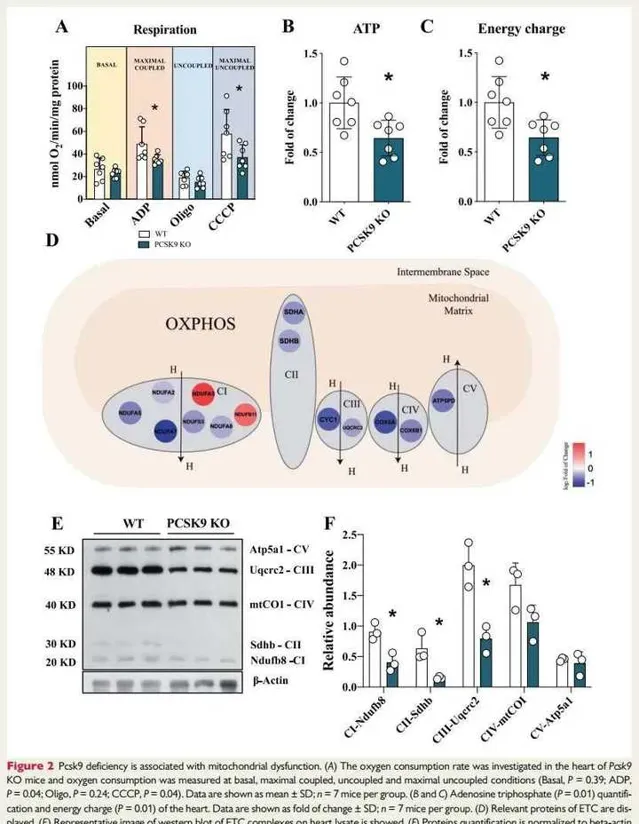
图2:Pcsk9缺乏与线粒体功能障碍有关。
图2A:在Pcsk9 KO小鼠的心脏中研究耗氧率,并分别在基础( Basal )、最大耦合(ADP)、非耦合(Oligo)和最大非耦合(CCCP)条件下测量耗氧量。结果发现:在最大耦合和最大非耦合呼吸条件下,与WT小鼠相比,Pcsk9 KO小鼠新鲜分离的心脏中表现出氧消耗率的显著降低。
图2B和C:为ATP定量和心脏的ATP能量电荷定量。数据显示为变化倍数。结果提示:与WT同窝对照以及其他参与能量产生的辅助因子相比,Pcsk9 KO小鼠心脏中的ATP水平(图2B)和ATP能量电荷(图2C)降低。
图2D:显示了ETC相关的蛋白质。与WT相比,Pcsk9 KO小鼠心脏中ETC复合物的关键结构成分蛋白质表达下调。
图2E:显示了ETC复合物的蛋白质印迹图像。
图2F:为蛋白质量化(对β-肌动蛋白表达的相比丰度)。结果提示:通过对代表每个线粒体复合物的蛋白质(包括NADH泛醌氧化还原酶亚B8(Ndufb8-复合物)、琥珀酸脱氢酶复合物亚基B(Sdhb-复合物Ⅱ)和泛醇﹣细胞色素 C 还原酶核心蛋白2(Uqcrc2-complexⅢ))进行蛋白质印迹分析,发现与WT同窝对照相比,在Pcsk9 KO小鼠的心脏中上述蛋白表达显著减少。综上,Pcsk9 KO小鼠的心脏中线粒体功能和含量受到影响。
我们接下来研究这是否会转化为心脏代谢的改变。Pcsk9 KO和WT小鼠心脏中代谢组学、蛋白质组学和脂质组学特征的综合分析(图3A和在线补充材料,图S3)显示线粒体活性受损的原因是:
①FA氧化受损——携带中链FA(C8和C10)的酰基肉碱的增加与β-氧化酶的减少,如甾醇载体蛋白2(SCP2)、烯酰辅酶A水合酶和短链1(ECHS1)(图3A和在线补充材料,图S3C和D);
②降低三羧酸(TCA)循环通量减少——TCA中间体和催化关键酶的酶水平降低(图3A和在线补充材料,图S3A和E)。
葡萄糖-6P水平的降低(图3A和在线补充材料,图S3B和F)和乳酸脱氢酶水平的增加以及乳酸血浆水平的增加(图3A)进一步证实了向无氧代谢的转变。然而,这不足以支持心脏能量需求;的确,我们观察到与WT相比,Pcsk9 KO小鼠心脏中能量电荷净减少(图2C)。
一致地,心脏蛋白质组的功能通路分析显示,与无法维持细胞能量需求相关的心脏功能障碍增加(图3B)。线粒体电子显微镜分析WT和Pcsk9 KO心脏,发现与WT相比,Pcsk9 KO心脏中心肌细胞中线粒体嵴的密度和组织性较低(图3C)。
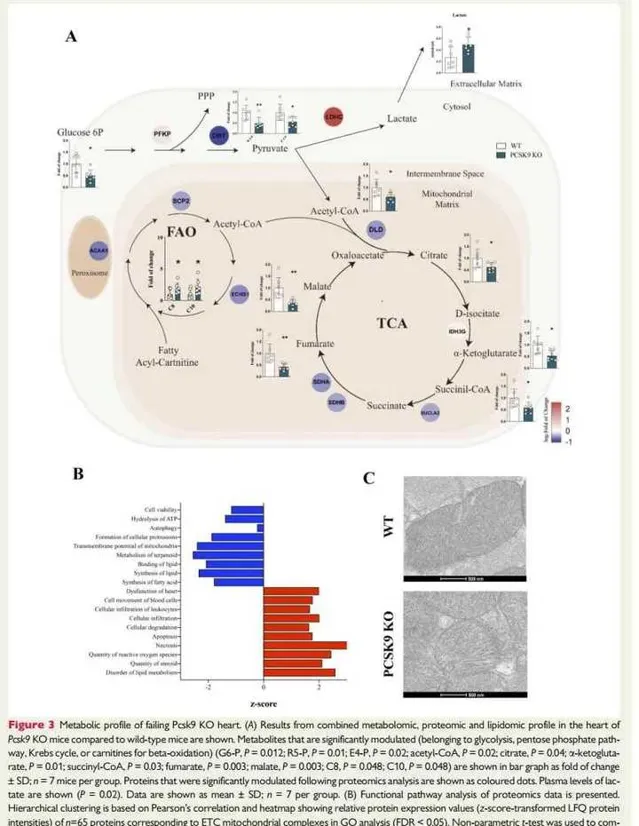
图3:Pcsk9 KO小鼠衰竭心脏的代谢特征。
图3A:显示WT和Pcsk9 KO小鼠心脏中组合代谢组学、蛋白质组学和脂质组学特征的综合分析。蛋白质组学分析后显著调节的蛋白质显示为彩色点。结果显示:显示线粒体活性受损的原因是:
①FA氧化受损——携带中链FA(C8和C10)的酰基肉碱的增加与β-氧化酶的减少,如甾醇载体蛋白2(SCP2)、烯酰辅酶A水合酶和短链1(ECHS1);
②降低三羧酸(TCA)循环通量减少——TCA中间体和催化关键酶的酶水平降低。葡萄糖-6P水平的降低和乳酸脱氢酶水平的增加以及乳酸血浆水平的增加进一步证实了向无氧代谢的转变。
图3B:显示了蛋白质组学数据的功能通路分析。分层聚类基于Pearson相关性和热图,显示了与GO分析中ETC线粒体复合物相对应的65种蛋白质的相对蛋白质表达值(z分数转换的LFQ蛋白质强度)。结果显示:无法维持细胞能量需求相关的心脏功能障碍增加。
图3C:显示了通过透射电子显微镜获得的心肌线粒体的代表性显微照片。结果发现:与WT相比,Pcsk9 KO心脏中心肌细胞中线粒体嵴的密度和组织性较低。
PCSK9缺乏导致心脏中LDLR和CD36表达增加以及脂质累积
为了进一步了解心脏功能障碍表型背后的分子机制,我们研究了心脏中PCSK9关键靶标的表达。与WT同窝对照相比,Pcsk9 KO小鼠中LDLR和CD36的表达均增加(图4A和 B )。值得注意的是,心脏的透射电子显微镜分析显示WT和Pcsk9 KO小鼠中纵向肌原纤维和规则插入的闰盘图(4C和D)。
然而,Pcsk9 KO心脏呈现出丰富的与线粒体紧密相联的脂滴(图4D),这一特征在WT小鼠中几乎不存在(图4C)。这一观察结果进一步支持了Pcsk9 KO小鼠心脏中脂质累积的存在。超薄切片的定量分析表明,Pcsk9 KO小鼠心脏中的脂滴数量(图4E和在线补充材料,图S4A)以及脂滴直径(图4F)均显著增加。
此外,与WT同窝对照相比,在Pcsk9 KO小鼠心脏中观察到总胆固醇(图4G)和花生四烯酸水平(在线补充材料,图S4B)增加,但总甘油三酯含量相似(图4H)。正如预期的那样,与WT小鼠相比,Pcsk9 KO小鼠的血浆胆固醇和甘油三酯水平较低(在线补充材料,图S3G和H)。
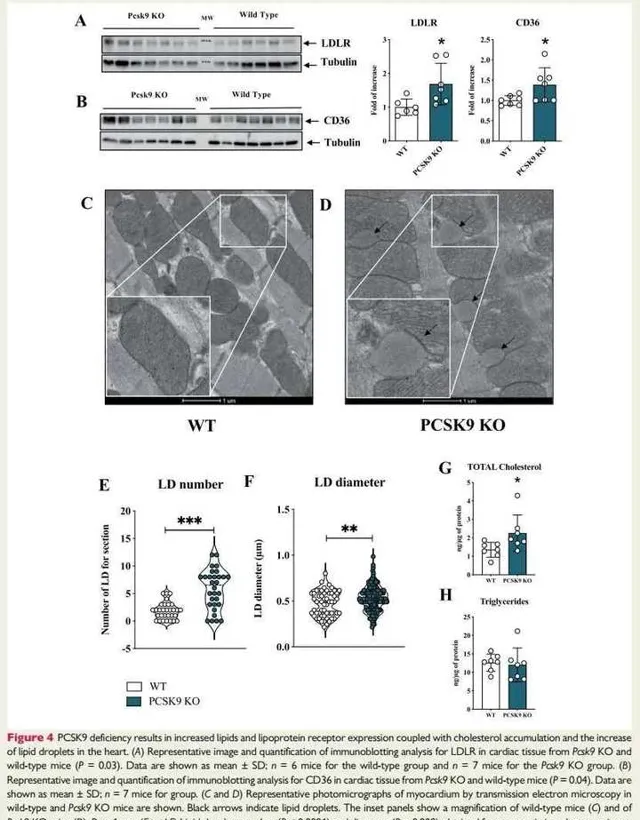
图4:PCSK9缺乏导致脂质和脂蛋白受体表达增加,并伴有胆固醇积累和心脏中脂滴的増加。
图4A:Pcsk9 KO和WT小鼠心脏组织中LDLR免疫印迹分析的代表性图像和量化结果。结果提示:与WT同窝对照相比,Pcsk9 KO小鼠中LDLR表达增加。
图4B:为Pcsk9 KO和WT小鼠心脏组织中CD36免疫印迹分析的代表性图像和量化结果。结果显示:与WT同窝对照相比,Pcsk9 KO小鼠中CD36表达增加。
图4C和D:显示了在WT和Pcsk9 KO小鼠中通过透射电子显微镜观察心肌的代表性显微照片。黑色箭头表示脂滴。结果提示:心脏的透射电子显微镜分析显示WT和Pcsk9 KO小鼠中纵向肌原纤维和规则插入的闰盘。然而,Pcsk9 KO心脏呈现出丰富的与线粒体紧密相联的脂滴,这一特征在WT小鼠中几乎不存在。这一观察结果进一步支持了Pcsk9 KO小鼠心脏中脂质累积的存在。
图4E和F:为透射电子显微镜分析中获得的脂滴数和直径。结果提示:Pcsk9 KO小鼠心脏中的脂滴数量以及脂滴直径均显著增加。
图4G和H:分别显示了心内总胆固醇和甘油三酯水平。结果提示:与WT同窝对照相比,在Pcsk9 KO 小命心脏中观察到总胆固醇(图4G)增加,但总甘油三酯含量相似(图4H)。PCSK9缺乏导致心脏中LDLR和CD36表达增加以及脂质积累。
观察到脂滴数量和胆固醇含量增加,加上Pcsk9 KO小鼠心脏中LDLR受体表达增加,表明LDLR在促进心脏脂质累积方面可能发挥作用。为了验证这一假设,我们对缺乏PCSK9和LDLR(DKO)小鼠的心脏功能、形态和代谢进行了表征。
与Ldlr KO小鼠相比,DKO小鼠在疲劳测试中观察到的跑步距离和时间显著减少(图5A和B);这种效果不依赖于骨骼肌力量的差异,因为前肢抓地力的测试结果相似5C)。
值得注意的是,与WT相比,Ldlr KO小鼠的心脏左心室质量/重量和收缩期心脏LVPW厚度显著降低(左心室质量/重量:WT 5.31±0.98与Ldlr KO 4.10±0.91,P =0.04)( LVPW:WT 1.23±0.13 Vs . Ldlr KO 1.10±0.08,P=0.04)(在线补充材料,表S1),这一发现与他们跑步耐量较差(与 WT 相比)一致(跑步距离:WT 331.1±46.20与 Ldlr KO 245.3±40.93m,P=0.049)(补充材料在线,图S5A-C)。
另一方面, DKO和Pcsk9 KO小鼠超声心动图观察到的特征非常相似(图1B和5C),表明PCSK9对心脏功能的影响不依赖于LDLR表达的调节和血浆胆固醇水平的变化(在线补充材料,图S5D和 E )。事实上,超声心动图分析表明,与 Ldlr KO小鼠相比, DKO小鼠在收缩期(图5D)表现出左心室显著增厚,而射血分数没有改变(图5E)。
此外,代谢分析表明,与Ldlr KO小鼠相比, DKO小鼠的ATP水平和能量电荷仍显著降低(图5F和G)。该曲线与TCA循环流量减少以及心脏线粒体功能障碍(图5H)以及血浆乳酸水平升高一致(图51和在线补充材料,图S5F)。这些数据强烈支持这样一个概念,即PCSK9缺乏引起的心脏LDLR表达增加与观察到的表型无关。
为了进一步了解PCSK9与心肌细胞生理学之间的联系,我们对从人类多能干细胞(iPSC-CM)中分化而来不同的心肌细胞进行了一系列研究。心肌细胞表型的确认通过肌钙蛋白T表达的增加(高达300倍)来证明(在线补充材料,图S6A)。
与在对照培养基中生长的细胞相比,补充VLDL导致PSC-CM中LDLR 、CD36、 LPL、4型葡萄糖转运蛋白(GLUT4)禾和 FA 合酶的mRNA表达显著增加(在线充材料,图S6B)。此外,VLDL处理导致线粒体质量减少(在线补充材料,图S6C)和中性脂质累积增加(在线补充材料,图S6D)。
当用PCSK9(在线补充材料,图S6E和 F )对心肌细胞进行预处理时,这些效应被逆转,证实了PCSK9在调节心肌细胞脂质摄取和线粒体功能中的作用。与这些发现一致,与WT相比,VLDL处理的Pcsk9 KO小鼠心脏分离的原代心肌细胞中显示线粒体质量减少,对于分离出的Pcsk9/Ldlr DKO的原代心肌细胞也是如此(在线补充材料,图S6G),因此进一步排除了观察表型中的LDLR的作用。
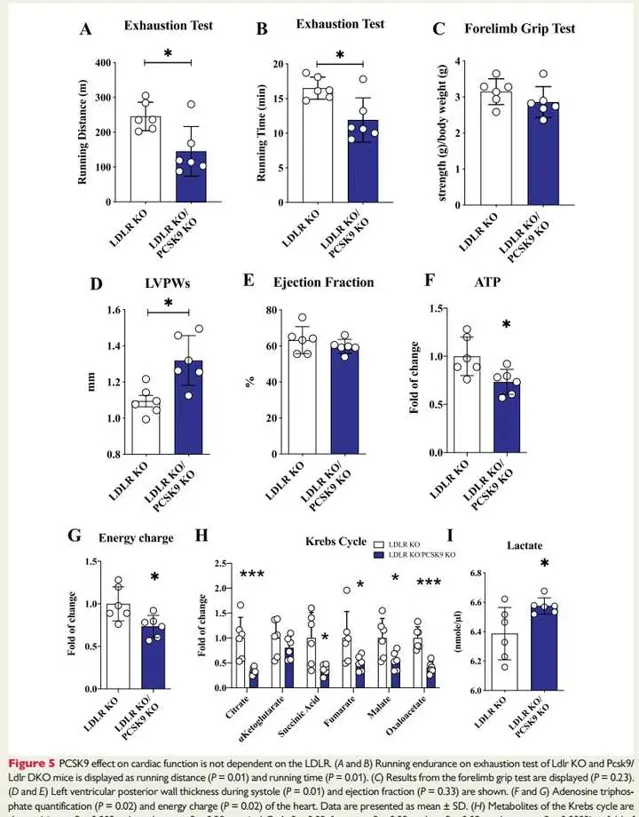
图5:PCSK9对心脏功能的影响不依赖于LDLR。
图5A和B:为Ldlr KO和Pcsk9/Ldlr DKO小鼠疲劳测试:分别为跑步距离和跑步时间。结果提示:与Ldlr KO小鼠相比, DKO小鼠在疲劳测试中观察到的跑步距离和时间显著减少。
图C:显示前肢抓地力测试结果,提示DKO小鼠的运动耐量降低不依赖于骨骼肌力量的差异,因为前肢抓地力的测试结果与Ldlr KO小鼠相似。
图5D和E:显示收缩期左心室后壁厚度和射血分数。结果提示:与Ldlr KO小鼠相比, DKO小鼠在收缩期表现出左心室显著增厚(D),而射血分数没有改变(E)。
图5F和G:为心脏的 ATP 定量和能量电荷。结果提示:与Ldlr KO小鼠相比, DKO小鼠的 ATP 水平和能量电荷均显著降低。
图5H:显示TCA循环的代谢物(柠檬酸盐,α-酮戊二酸,琥珀酰辅酶A,富马酸,苹果酸,草酰乙酸)的变化。结果提示TCA循环流量减少以及心脏线粒体功能障碍。图5显示乳酸的血浆水平。结果提示:与 Ldlr KO小鼠相比,DKO小鼠的血浆乳酸水平升高。综上,PCSK9对心脏功能的影响不依赖于LDLR。
循环PCSK9不影响心脏代谢
在人类和小鼠中,循环PCSK9主要来源于肝脏。因此,为了区分循环与局部产生的PCSK9对心脏功能的影响,我们分析肝脏中PCSK9选择性缺乏的小鼠(AlbCre +/Pcsk9LoxP/LoxP小鼠,仅缺乏循环PCSK9)的心脏功能和心脏形态。与 AlbCre-/Pcsk9LoxP/LoxP小鼠相比,AlbCre+/Pcsk9LoxP/LoxP小鼠在疲劳测试中观察到的跑步距离和跑步时间相似(图6A和B),肌肉表现也相似(图6C)。
因此,收缩期左心室厚度(图6D)、射血分数(图6E)以及几个心脏参数(在线补充材料,图S7A-E)没有出现差异。此外,ATP产生(图6F)、ATP能量电荷(图6G)、三羧酸循环代谢物(图6H)和糖酵解中间体(在线补充材料,图S7F)在这些小鼠的心脏中没有差异。
与AlbCre-/Pcsk9LoxP/LoxP对照小鼠相比,AlbCre+/Pcsk9LoxP/LoxP小鼠的循环乳酸水平(图6J)和甘油三酯(在线补充材料,图S7G)未受影响,而肝脏选择性KO小鼠中的胆固醇显著降低(在线补充材料,图S7H)。 这些数据排除了在观察表型中肝脏产生(即循环)PCSK9缺乏的作用,提示局部产生的PCSK9缺乏是心脏功能障碍的驱动因素。
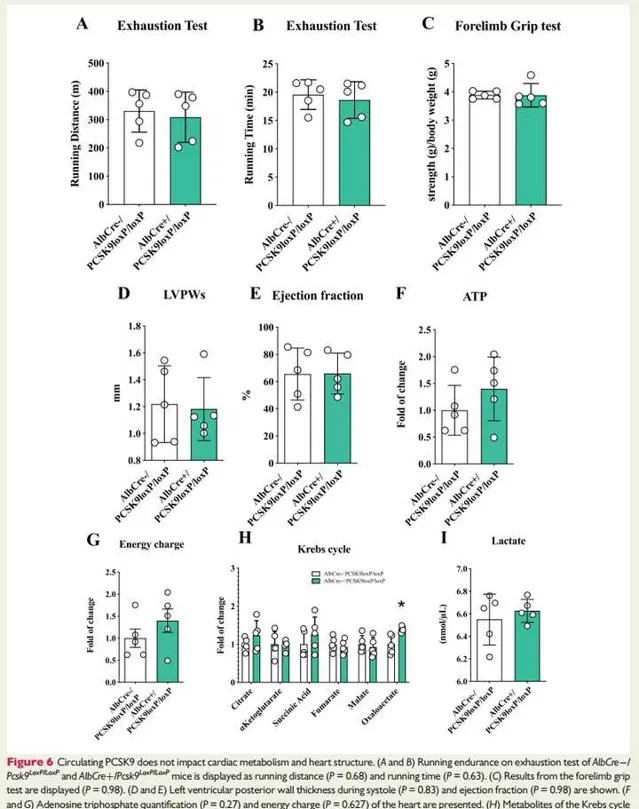
图6:循环PCSK9不会影响心脏代谢和心脏结构。
图6A和B:为 AlbCre -/Pcsk9 LoxP/LoxP和AlbCre +/Pcsk9 LoxP/LoxP小鼠疲劳测试:分别为跑步距离和跑步时间。结果提示:与 AlbCre-/Pcsk9LoxP/LoxP 小鼠与AlbCre +/Pcsk9LoxP/LoxP 小鼠在疲劳测试中观察到的跑步距离和跑步时间 相似。
图6C:显示前肢抓地力测试。结果提示:AlbCre -/Pcsk9LoxP/LoxP 小鼠和 鼠骨骼肌机能相似。
图6D和E:显示收缩期左心室后壁厚度和射血分数。结果发现:AlbCre -/Pcsk9LoxP/LoxP 小鼠和 AlbCre +/Pcsk9LoxP/LoxP 小鼠收缩期左心室厚度(图6D)、射血分数(图6E)没有明显差异。
图6F和G:显示了心脏的ATP定量和能量电荷。
图6H:为TCA的代谢物(柠檬酸,α-酮戊二酸,琥珀酰辅酶A,富马酸,苹果酸,草酰乙酸)变化。ATP产生(图6F)、ATP能量电荷(图6G)、三羧酸循环代谢物(图6H)在这些小鼠的心脏中没有差异。
图6I:显示了乳酸的血浆水平。与 AlbCre-/Pcsk9LoxP/LoxP 对照小鼠相比, AlbCre+/Pcsk9LoxP/LoxP 小鼠的循环乳酸水平未受响。综上,肝脏产生(即循环)PCSK9缺乏对心脏功能无明显影响,提示局部产生的PCSK9缺乏是心脏功能障碍的驱动因素。
遗传性PCSK9功能丧失与人类心脏表型的改变有关
为了进一步转化我们在人类中的发现,我们评估了PCSK9功能丧失变异(R46L)对来自普通人群的2606名受试者的基于超声心动图的心脏功能标志物的影响(PLIC研究)。对12名杂合R46L携带者的心脏特征进行初步分析(在线补充材料,表S2)显示,与年龄和性别匹配的WT受试者相比,这些受试者的左心室质量指数显著增加(图7A),但射血分数相似(图7B)。有趣的是,杂合子受试者和WT携带者的腿部和手臂骨骼肌质量相当(图7C)。
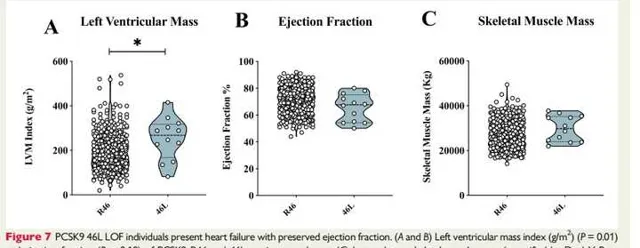
图7:PCSK9 46L LOF个体出现射血分数保留的心力衰竭。
图7A和B:显示了PCSK9 R46和46L携带者的左心室质量指数(g/m²)和射血分数。结果提示:与年龄和性别匹配的WT受试者相比,这些受试者的左心室质量指数显著增加(图7A),但射血分数相似(图7B)。
图7C:显示腿部和手臂骨骼肌质量(通过双X射线吸收法量化,遵循汉森公式)。结果提示:杂合子受试者和WT携带者的腿部和手臂骨骼肌质量相当。
讨论
在这项研究中,我们证明PCSK9在控制心脏代谢和功能方面起着关键作用。当PCSK9缺失时,参与脂质和脂蛋白摄取的关键受体的表达增加,导致心脏胆固醇积累、β-氧化和线粒体活性受损,从而影响心脏代谢和功能(图文摘要)。
与肝脏不同,心脏不能合成大量FA,因此脂质需求主要通过从循环中摄取来满足,但是需要适当控制以限制过量摄取(可能导致脂质累积和细胞脂毒性)。在这种情况下,心肌细胞将其代谢转向无氧糖酵解,但这并不能完全补偿增高的能量需求。
从生理上讲,这种转变会触发心脏的一系列形态适应,包括增加左心室壁厚度以维持射血分数。这种特征是HFpEF的典型特征,其中心肌细胞将其代谢从FA氧化转为线粒体,然后转换为糖酵解和酮体利用,如高血压和肥胖以及糖尿病患者中所描述的情况。
本研究表明在实验模型的PCSK9缺陷条件下观察到类似的特征。心脏代谢特征的详细分析表明,耗氧率和ATP水平降低是线粒体功能受损的结果,同时ETC蛋白复合物和活性的改变也提示了这一点。
先前的观察突出心脏脂质累积会导致脂毒性并促进心脏代谢转换;因此,PCSK9缺乏导致心脏中胆固醇累积增加以及代谢转向无氧糖酵解的观察结果支持「PCSK9在维持与心肌细胞脂质摄取相关的因素适当平衡方面发挥生理作用」这一假设。
与这种活性明显相关的候选因子就是脂蛋白受体。尽管与其他组织(如肝脏)相比,其在心脏中的表达程度较低,但仍在控制心脏脂蛋白摄取和脂质代谢方面发挥关键作用。LDL 和CD3这两个PCSK9靶标的表达在Pcsk9 KO小鼠的心脏中增加可能代表了接受PCSK9抑制剂治疗的患者关注的问题,因为LDLR再循环的增加是导致肝脏中脂蛋白摄取增加和血浆胆固醇降低的原因,但也可能影响心脏脂质累积。
为了进一步阐明这方面,在Pcsk9/Ldlr DKO小鼠和Ldlr KO小鼠中研究了心脏特征和跑步耐量。值得注意的是,Pcsk9/Ldlr DKO小鼠仍然存在心脏功能障碍和在Pcsk9 KO小鼠中观察到的代谢后遗症,而Ldlr KO与WT相比,收缩期的左心室质量/重量和心脏LVPW厚度显著降低,因此排除了PCSK9观察到的心脏表型中LDLR轴的作用。此外,这一发现表明,至少在心脏中,LDLR介导的脂蛋白摄取最终不会导致脂质累积和心脏代谢损伤。
PCSK9的药理学抑制是否会导致心脏脂质摄取增加和心脏功能障碍吗?来自PCSK9抑制剂的大型介入性试验数据并未报告心力衰竭的发生率增加。
目前针对PCSK9的疗法包括通过隔离循环PCSK9起作用的单克隆抗体,以及通过选择性沉默肝脏中PCSK9 mRNA表达的基因沉默方法。鉴于肝脏影响循环PCSK9水平,这两种方法都会减少循环PCSK9,但不影响局部(除外肝脏)PCSK9的产生。
为了探索完全缺乏循环PCSK9是否会影响心脏代谢,在肝脏中PCSK9表达选择性缺乏的实验模型中评估了心脏功能:循环PCSK9水平低于检测水平,但在其他组织中的表达未改变。这是一种模仿PCSK9抑制剂治疗后观察到的表型。
在这个实验模型中,运动表现、心脏形态、代谢特征和耗氧量与WT小鼠均相似。这一观察结果对于排除循环PCSK9缺乏对心脏代谢的影响具有开创性意义,并且与抗PCSK9疗法临床试验中观察到的心脏功能数据一致。
在基础条件下,PCSK9在心脏中的表达水平非常低(在线补充材料,图S71),但在缺血条件下(体内)和与氧化LDL孵育后(体外),其水平被诱导性升高。据推测,缺血/再灌注损伤后立即诱导PCSK9可能通过刺激自噬过程和去除受损线粒体来保护急性期的心脏,这种作用随着时间的推移可能变得有害,可能导致细胞破坏和心肌细胞死亡增加。心外膜脂肪组织局部产生的PCSK9也有可能导致了这种病理生理变化,这一发现得到了R46L变异与人类心外膜脂肪累积增加相关的观察结果的支持(与肥胖或糖尿病无关)。然而,我们没有发现心外膜脂肪厚度与左心室质量之间存在任何相关性(数据未显示),支持PCSK9对心外膜脂肪组织炎症影响主要与射血分数降低的心力衰竭有关这一假说。
未来在更大的队列中开展研究应该解决PCSK9功能丧失变异是否与心脏表型改变相关,而与在这些情况下观察到的LDL-胆固醇水平的长期降低所导致的较低心血管风险无关。我们必须承认,在完整的Pcsk9 KO小鼠和肝脏选择性PCSK9 KO模型中分析了心脏功能。虽然排除了循环PCSK9对观察表型的作用,但应在心脏选择性KO模型中对PCSK9心脏的选择性作用进行最终确认。
同样,Pcsk9/CD36 DKO的表征将有助于研究心脏应激后PCSK9的产生是否可能代表一种反馈机制,有助于维持心脏脂质摄取和脂质累积之间的适当平衡,从而限制心脏脂毒性的潜在副作用。










