更新:6/24/2020 2:10AM
謝 @Antioque 提醒,之前的回答裏我發現確實只考慮了一種情況(中性受質和帶負電的親核基團)而沒有繼續分析其他情況,現在我在根據以往學過的基礎知識加上 @Antioque 的提醒我重新梳理了一下溶劑極性對E1、E2、SN1、SN2反應的影響進行一次簡單的定性分析。我簡單的歸納一下,就是根據相似相溶原理, 如果反應物到過渡態活化分子的過程中極性增強,那麽相應的溶劑的極性增強將加快反應速率。反之,如果反應物到過渡態活化分子的過程中極性減少,那麽相應的溶劑的極性減少將加快反應速率 。極性,字面意思,部份偶極矩增大,所以極性的大小與電場強度的分布均勻性呈負相關。
補充說明:
在SN1中,受質的離去基團的離去是決速步驟,如果是中性受質 \text{R}\text{—}\text{LG} ,那麽離去基團與中性受質之間的化學鍵斷裂時電荷分布會呈現 \text{R}^{\delta+}\text{------}\text{LG}^{\delta-} ,由於碳正離子 \text{R}^{+} 的點正電荷增大並且離去基團 \text{LG} 的點負電荷也增大,導致電場強度分布更不均勻,即極性增大。如果是帶正電的受質 \text{R}\text{—}\text{LG}^{+} ,那麽離去基團與中性受質之間的化學鍵斷裂時電荷分布會呈現 \text{R}^{\delta+}\text{------}\text{LG}^{\delta+} ,由於碳正離子 \text{R}^{+} 的點正電荷增大而離去基團 \text{LG} 的點正電荷減弱,導致電場強度分布更均勻,即極性減小。而極性溶劑中質子化溶劑又比非質子化溶劑極性更強,使得中性受質的解離能顯著性降低,如果受質比作一張紙,那麽溶劑的極性大小就是雙手撕扯紙張的強弱。例如水 \text{H}_{2}\text{O} 可以微弱自解離成 \text{H}_{3}\text{O}^{+} 和 \text{OH}^{-} ,然後誘導效應異性相吸使得反應物從初態 \text{OH}^{-}\text{------}\text{R}\text{—}\text{LG}\text{------}\text{H}_{3}\text{O}^{+} 到過渡態 \text{OH}^{-}\text{---}\text{R}^{\delta+}\text{------}\text{LG}^{\delta-}\text{---}\text{H}_{3}\text{O}^{+} 相比於非質子化和非極性溶劑所需的解離能更低。對於帶正電的受質 \text{R}\text{—}\text{LG}^{+} 則相反,由於極性反轉,即反應物初態變成 \text{H}_{3}\text{O}^{+}\text{------}\text{R}\text{—}\text{LG}^{+}\text{------}\text{OH}^{-} 根據異性相吸原理,導致過渡態 \text{H}_{3}\text{O}^{+}\text{---}\text{R}^{\delta+}\text{------}\text{LG}^{\delta+}\text{---} \text{OH}^{-} 中的電場分布相比於初態更不均勻,即更不穩定。所需的活化能也就相比於弱極性或非極性溶劑環境下更高。如果把受質比作被壓縮的彈簧,那麽極性分子就是那雙摁住彈簧的手。
在SN2中,由於在動力學下是一步反應,所以親核基團和受質的活性共同決定反應速率,根據電性一共有2*2=4種情況
- 中性受質和負電性親核基團 \text{Nu}^{-}\text{------}\text{R}\text{—}\text{LG}\rightarrow \text{Nu}^{\delta-}\text{---}\text{R}\text{------}\text{LG}^{\delta-}
- 中性受質和中性親核基團 \text{Nu}\text{------}\text{R}\text{—}\text{LG}\rightarrow \text{Nu}^{\delta+}\text{---}\text{R}\text{------}\text{LG}^{\delta-}
- 正電性受質和負電性親核基團 \text{Nu}^{-}\text{------}\text{R}\text{—}\text{LG}^{+}\rightarrow \text{Nu}^{\delta-}\text{---}\text{R}\text{------}\text{LG}^{\delta+}
- 正電性受質和中性親核基團 \text{Nu}\text{------}\text{R}\text{—}\text{LG}^{+}\rightarrow \text{Nu}^{\delta+}\text{---}\text{R}\text{------}\text{LG}^{\delta+}
第一、三和四種情況裏電場強度分布更均勻,即極性減少。而第二種則相反,電場強度分布更不均勻,即極性增大。所以對於第一、三、四種情況,減少溶劑的極性有助於加快反應速率。對於第二種情況,增加溶劑的極性有助於加快反應速率。
E1和E2與SN1和SN2基本同理,其中有區別的地方是E2和SN2中使用的溶劑和親核試劑(Lewis堿)的交互作用會影響Lewis堿的親核性。質子化溶劑均會抑制中性和負電性親核試劑的親核性,因為親核試劑作為Lewis堿,即給電子體會使得該親核試劑的偏負電部份會與溶劑的偏正電部份——氫原子形成氫鍵,導致親核試劑與質子化溶劑配合或因高位阻效應而失去親核性,所以抑制了SN2反應。當然也會抑制SN1反應但是相比SN2,SN1反應要求的Lewis堿不需要那麽強的親核性。溫度也是一個重要的因素,通常來講消去反應相對高溫。
以上就是現階段我已經回顧的大二有機化學一部份基礎知識了,如果有什麽遺漏、錯誤和不嚴謹的地方也請各位讀者在評論區留言。最近本人在專註於數學和電腦選修,以便以後跨專業領域學習與工作。
原回答
題主首先得了解這四種反應的機制才能了解具體在什麽條件下哪種反應占主導(酸堿性、極性、溫度、位阻效應、活化配合物穩定性、手性等等等等)。我看了題目的簡介,題主是想著重分析溶劑極性的影響,那麽此回答我就著重分析極性,當然要記住還有其他因素會影響這四種反應之間的競爭。以下是我對大二時期學的有機化學知識的回顧。總的來說
E1是單分子消去反應,先是受質中的離去基團被解離並且在原中心碳位置上形成碳正離子,然後鄰位β碳原子上的氫被拔去後形成π鍵,該反應在動力學上分為兩部,第一步是速率決定步驟,所以受質的活性影響反應速率。
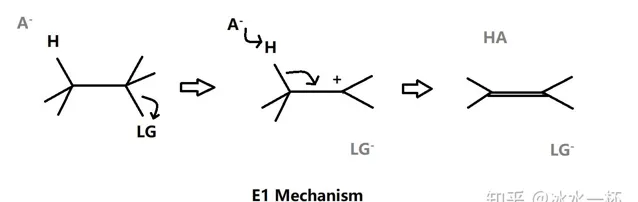
該反應在極性溶劑條件下更有利,這是因為離去基團的解離並形成碳正離子是決速步驟,形成的碳正離子會與極性溶劑分子的偏負電荷部碎形成離子偶極矩分散電荷密度從而保持穩定(就像食鹽水溶液中的鈉離子和水分子中的偏負電荷部份——氧原子上的交互作用),即溶劑化效應。質子極性溶劑穩定作用更強,那是因為形成的離子偶極矩是氫鍵,氫鍵比大多數分子間交互作用強。所以該反應非常適合用酸催化。因為在第二步sp3混成碳原子上的C-H鍵斷裂時需要相應的活化能,所以E1反應通常在相對高溫的環境下進行,否則會發生SN1反應而不是E1反應(詳細見下文SN1反應)。同時,溫度的上升也會加強溶劑化效應,這是因為溫度的上升使得溶劑分子的自解離反應平衡向解離方向移動從而增強溶劑的極性。
E2是雙分子消去反應,Lewis堿直接進攻拔去鄰位β碳原子上的氫同時對位上的離去基團被解離從而形成π鍵,該反應在動力學上只有一步,受質和Lewis堿的活性共同影響反應速率

該反應在非極性溶劑條件下更有利,這是因為Lewis堿中的偏負電荷部份會與極性溶劑分子的偏正電荷部碎形成離子偶極矩分散電荷密度從而保持穩定(就像食鹽水溶液中的氯離子和水分子中的偏正電荷部份——氫原子上的交互作用),即溶劑化效應。質子極性溶劑分子甚至會直接與Lewis堿配合從而失去反應活性。所以該反應的發生需要強Lewis堿和高位阻效應(詳細見下文SN2反應)同時,溫度的上升也會加強溶劑化效應,從而在熱力學上減弱親核基團的親核性並且增強鄰位碳上的氫的解離性(達到解離所需的活化能)。所以在實驗室裏一般E2反應是在相對高溫的環境下進行。
SN1是單分子取代反應,先是受質中的離去基團被解離並且在原中心碳位置上形成碳正離子,然後親核基團進攻碳正離子形成新的σ鍵,該反應在動力學上分為兩部,第一步是速率決定步驟,所以受質的活性影響反應速率。
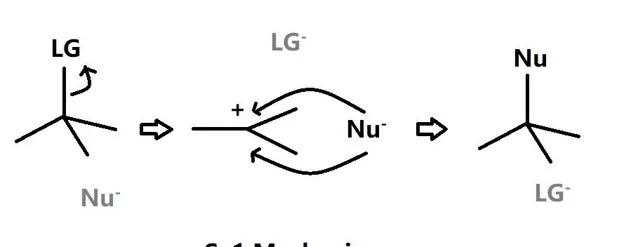
由此可以看出,與E1反應類似,該反應在極性溶劑條件下更有利,但是要註意,溫度的上升也會促進生成產物中的親核基團的解離性,從而在熱力學上降低生成產物的穩定性從而可能發生E1反應而不是SN1反應,即存在E1和SN1反應的天然競爭。
回顧下乙醇在濃硫酸的催化下脫水生成乙烯的E1反應 \text{EtOH}\xrightarrow[170°\text{C}]{\text{H}_{2}\text{SO}_{4}} \text{C}_{2}\text{H}_{4}+\text{H}_{2}\text{O} 中的機理,第一步是決速步驟,即乙醇在酸性條件下脫水形成乙基正離子 \text{EtOH}+\text{H}^{+}\rightarrow\text{C}_{2}\text{H}_{5}^{+}+\text{H}_{2}\text{O} ,但是在第二步中,如果反應條件是在常溫條件下,即不滿足170度的高溫,那麽形成的碳正離子會直接被親核基團硫酸氫根離子進攻從而發生SN1反應並形成硫酸氫乙酯(單乙基硫酸) \text{C}_{2}\text{H}_{5}^{+}+\text{H}\text{SO}_{4}^{-}\rightarrow \text{EtHSO}_{4} 。 隨著溫度的上升,硫酸氫乙酯開始解離,在140度左右,由於在熱力學上沒有達到解離乙基正離子中sp3混成的碳上的氫所需的活化能,另一個乙醇分子上的氧上的一對孤電子會進攻已經解離出的乙基正離子然後被硫酸氫根離子拔去額外的氫從而生成乙醚 \text{EtOH}+\text{C}_{2}\text{H}_{5}^{+}+\text{HSO}_{4}^{-}\rightarrow \text{EtOEt}+\text{H}_{2}\text{SO}_{4} 。在170度時才會發生E1反應,乙基正離子上的鄰位碳上的氫被拔去並形成π鍵從而生成乙烯 \text{C}_{2}\text{H}_{5}^{+}+\text{HSO}_{4}^{-}\rightarrow \text{C}_{2}\text{H}_{4}+\text{H}_{2}\text{SO}_{4} 。
SN2是雙分子取代反應,親核基團直接進攻偏正電荷的碳同時對位上的離去基團被解離從而形成新的σ鍵,該反應在動力學上只有一步,受質和親核基團的活性共同影響反應速率。
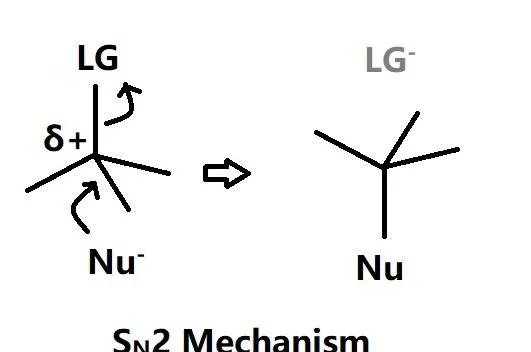
與E2反應類似,該反應在非極性溶劑條件下更有利,該反應的發生需要強Lewis堿和低位阻效應(例如小分子強親核性Lewis堿乙氧基負離子 \text{EtO}^{-} )。如果是大分子弱親核性Lewis堿如叔丁氧基負離子 t\text{BuO}^{-} (叔丁醇 (\text{CH}_{3})_{3}\text{COH} 脫去羥基上的氫形成) 那麽就可能會發生E2反應,即Lewis堿不會進攻偏正電荷的碳而是拔去鄰位碳上的氫。即存在E2和SN2反應的天然競爭。同時,溫度的上升也會加強溶劑化效應,從而在熱力學上減弱親核基團的親核性並且增強鄰位碳上的氫的解離性(達到解離所需的活化能)。所以在實驗室裏一般SN2反應是在相對低溫的環境下進行。
總結
極性低溫——SN1
極性高溫——E1
非極性低溫——SN2
非極性高溫——E2
要記住,還有其他因素(區域選擇性,手性,共振結構等等等等)會影響這四種反應的競爭,這個總結是在其他因素等同的條件下說明的。




