消息一出,圈内震动不小。
"堪比 AlphaFold"、"前所未有的准确度"、"结构预测里程碑"、"生物界海啸级存在"等形容出现在诸多解读文章中。RNA 结构预测本身的确困难重重,这项研究的终极意义是什么?对学界的影响究竟有多大?
就此,生辉联系到了深圳湾实验室系统与物理生物学所资深研究员周耀旗。
他的研究方向聚焦在结构生物信息学,曾经多次在国际蛋白质结构预测和功能预测比赛中名列前茅。到目前为止共发表论文 200 余篇,引用 1 万多次。其课题组主要围绕着 RNA 和蛋白质的序列、结构及功能之间的关系以及生物高分子的应用开发等几方面进行科学研究。
周耀旗于 2000 年任纽约州立布法罗大学助理教授,2004 年升为终身副教授,2006年成为印第安纳大学信息学院和医学院终身正教授,2013-2021 年任澳大利亚格里菲斯大学糖组学研究所正教授。

" 这个模型所训练出来的实际上是一个 RNA 结构的评价函数 :给一个人工结构模型,预测这个模型与真实的结构相差有多远。以前的打分函数一般是根据物理原理来推导或者通过对 RNA 结构构像进行统计分析,而 ARES 模型的不同之处是通过深度学习来获得这样的打分函数 。"周耀旗告诉生辉。
"对于 RNA 预测本身来说只是一小步"
与DNA 不同,RNA 是单链折叠成的隆起、假结、发夹等多种多样的复杂三维结构,在满足不同的功能状态需求时,不同的折叠状态也可以转换。但已知的非冗余 RNA 结构仅不到 250 个,大多数RNA结构未知。
2019 年,斯坦福大学的生物化学副教授 Rhiju Das 博士发表了一篇文章, FARFAR2: Improved de novo Rosetta prediction of complex global RNA folds 。结果显示,在没有实验数据或者专家的干预下,FARFAR2 预测的 RNA 模型更接近与 RNA 的自然构象,优于其它参与 RNA-Puzzles 的小组提交的模型。
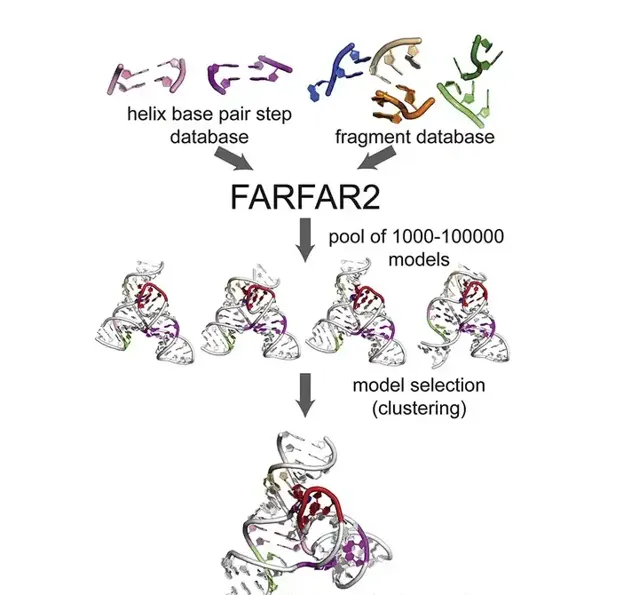
(来源: FARFAR2: Improved de novo Rosetta prediction of complex global RNA folds )
RNA-Puzzles 是由科学界组织的一项长期挑战,旨在解析 RNA 的三级结构。根据 RNA-Puzzles 的规则,当科学家们通过实验发现新的 RNA 结构时,他们不会公开细节,要等到其他 RNA-Puzzles 参与者提交了各自的结构预测,之后再把实验确定的结构与这些结构进行匹配程度的对比判断。
事实上, 研究团队这次仅仅是为他们之前开发的 RNA 预测软件 FARFAR2 开发了一个打分函数。
与 AI 直接预测结构不同,ARES 的框架并不是针对 RNA 的天然结构设计,而是针对人工模型与天然结构的比较,通过不断地调整参数,ARES 可以深入了解 RNA 人工模型与天然结构上每个原子之间的相对位置以及几何排列的不同,进而推算出人工模型与天然结构的差距有多远。
"以前也有人做过,但从文章上来看,该模型训练的鲁棒性比较好,也就是说应用在没见过的结构模型上,结果也不错。 这个模型的出现对于业内的最大影响可能是,进一步让大家认识到把深度学习应用到 RNA 结构预测上的优势, 这也是我们组近几年一直努力的方向。"
"但对RNA结构预测本身来说还只是一小步,因为RNA结构预测最难的部分是折叠成接近真实天然结构(near native structure)的构像,ARES 模型并没有涉及。也就是说,ARES 模型需要跟能够产生结构构像的 FARFAR2 结合才能预测 RNA 结构,而 FARFAR2 在多数情况下并不能产生很好的结构模型。 "周耀旗总结道。
值得注意的是,本文的通讯作者 Ron Dror 博士和 Rhiju Das 博士的研究团队曾在 2020 年共同在 Proteins 上发表了一篇文章,利用机器学习的方式,通过生物大分子中每个原子的位置,及原子之间的相对位置,识别蛋白质复合物的模型。本次也是继在蛋白质上的成功,继续探讨的研究成果。
"人为因素过多,效果未知"
AlphaFold 收集了数千种已知蛋白结构进行训练,与之不同,ARES 模型只用了 18 个已知的 RNA 结构组件,包括从 1994 年到 2006 年之间公布的数据,每个 RNA 结构组件用 Rosetta FARFAR2 生成了 1000 个结构模型。
值得注意的是,在训练 ARES 时,并未预先将结构模型的特征编程到系统中,即使是双螺旋、碱基对、核苷酸和氢键登基本概念也没有被预先编程到系统中,而是让算法自己去发现。这样做是因为如果为机器提供额外的"学习资料",会使算法偏向于选择某些特征,如此就会阻止它找到其他新特征。
那么,用 18 个已知的RNA结构训练出来的ARES模型是否可靠?
"这 18 个结构组件可以说包含了 RNA 已知结构的精要了,每个结构组件产生了 1000 个模型, 应该说 18×1000 的数据量还可以 ",周耀旗补充道,"已知的 RNA 结构很少,已知的 RNA 结构组件更少,所以这个模型能够从 18 个结构组件训练出来,也不完全是意外,毕竟对 ARES 模型的要求不是那么高,把比较好的结构(更接近真实结构)的模型排序高一点,而且它是针对 FARFAR2 方法预测的模型来排序的,因为 FARFAR2 方法预测出来的模型有一定的一致性,所以训练出来的鲁棒性比较有保证。"
但周耀旗也指出, 并不能保证用 ARES 模型去给其他方法预测出来的模型打分的效果会一样好。该模型现在仅仅是一个打分函数,里面存在太多的人为因素,比如,研究人员选择用 18 个 RNA 结构组件来训练模型。
"为什么是 18 个 RNA 结构组件来训练模型?如果用更多的 RNA 来训练,会有什么不同?"周耀旗也向大家抛出了一个问题。
尽管 AlphaFold 的出现成为蛋白质结构预测领域可喜的进展,但 RNA 的结构预测依然存在着很大的问题。
蛋白质有 20 种氨基酸,包括亲水、疏水、带电荷、不带电荷的,信息量很大,而 RNA 只有4种碱基,信息量少是 RNA 结构预测的难点之一。
此外,RNA 结构的稳定性低,受环境影响大,这也导致对于 RNA 结构的研究更加困难。
评分属同类最优?
为了评估 ARES 模型的准确性,研究人员使用了从 2010 年到 2017 年间发表的 RNA 结构。对于每个 RNA 结构,研究人员使用 FARFAR2 软件生成了至少 1,500 个结构模型。然后,他们应用经过训练的 ARES 为每个模型生成一个分数。同时还使用了三个评分函数,分别是Rosetta、RASP、以及 3dRNAscore。
以 Rosetta 为例,在 Rosetta 中评估一个模型的好坏,最直观的方法就是使用 Rosetta 的打分系统进行评估,也就是常说的能量函数。通过能量函数,能够预测在什么构象下,能量最低,RNA 处于最佳状态,原子间的相互作用达到最优。
研究团队用两种方式与其它三种评分函数进行了对比。在第一个基准测试上,ARES 的性能大大优于其他三个评分功能:比如 62% 的基准 RNA 的单一最佳评分结构模型接近原生,且均高于其他三个模型。
在第二个基准测试上,ARES 再次完成了所有评分功能。而且,ARES 的最佳评分结构模型的 RNA 的中位数均方根偏差值,明显低于其他评分函数。
研究人员称即使在数据量少的情况下也可以成功训练评价RNA结构的打分函数,他们希望 ARES 模型能够帮助科学家解释不同分子的工作原理,应用范围从基础生物学研究到药物设计。
下图可以看出,ARES 预测的 RNA 的结构模型远接近于真实模型(4.8Å),而由 Rosetta 预测的模型则和真实模型有较大偏差( 7.7 Å )。
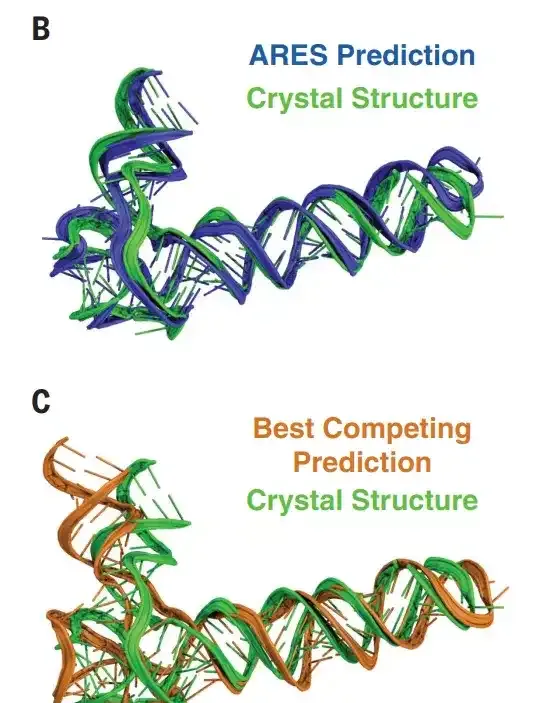
"FARFAR2 无疑是当今最好的方法之一,但目前的问题是,所有的方法都只能预测比较简单的 RNA 结构。"周耀旗说道。
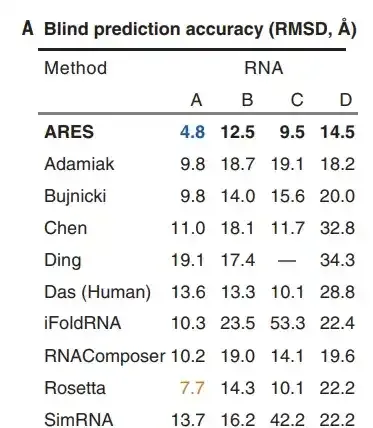
一般情况下,RNA 预测的结构至少<6 Å 才可能有点类似的模样。事实上,在盲测比赛中,ARES+FARFAR2 所预测的 4 个 RNA,除了一个是 4.8Å,其它都是 10-15Å。看起来好像比目前的方法好很多(10-20 Å, Bujnicki组),但其实大多数谁也不像。
周耀旗总结道,ARES 其实只是相当于 AlphaFold2 里面蛋白质人工结构评价的那部分,离 RNA 结构全预测有很大的距离。 毫无疑问,这个深度学习产生的 ARES 模型在RNA结构打分函数上迈出了可喜的一步,但这仅仅是 RNA 结构预测这个大问题里面的一个子问题。 这个子问题和其它子问题(例如二级结构预测、主链构像预测和结构高精度优化)的一起稳定进展才能为最终解决 RNA 结构预测问题作出贡献。
关于RNA更多相关讯息,在历史文章中搜索即可(gzh:生辉)。










