訊息一出,圈內震動不小。
"堪比 AlphaFold"、"前所未有的準確度"、"結構預測裏程碑"、"生物界海嘯級存在"等形容出現在諸多解讀文章中。RNA 結構預測本身的確困難重重,這項研究的終極意義是什麽?對學界的影響究竟有多大?
就此,生輝聯系到了深圳灣實驗室系統與物理生物學所資深研究員周耀旗。
他的研究方向聚焦在結構生物資訊學,曾經多次在國際蛋白質結構預測和功能預測比賽中名列前茅。到目前為止共發表論文 200 余篇,參照 1 萬多次。其課題組主要圍繞著 RNA 和蛋白質的序列、結構及功能之間的關系以及生物高分子的套用開發等幾方面進行科學研究。
周耀旗於 2000 年任紐約州立水牛城大學助理教授,2004 年升為終身副教授,2006年成為印第安納大學資訊學院和醫學院終身正教授,2013-2021 年任澳洲格裏菲斯大學糖組學研究所正教授。

" 這個模型所訓練出來的實際上是一個 RNA 結構的評價函數 :給一個人工結構模型,預測這個模型與真實的結構相差有多遠。以前的打分函數一般是根據物理原理來推導或者透過對 RNA 結構構像進行統計分析,而 ARES 模型的不同之處是透過深度學習來獲得這樣的打分函數 。"周耀旗告訴生輝。
"對於 RNA 預測本身來說只是一小步"
與DNA 不同,RNA 是單鏈折疊成的隆起、假結、發夾等多種多樣的復雜三維結構,在滿足不同的功能狀態需求時,不同的折疊狀態也可以轉換。但已知的非冗余 RNA 結構僅不到 250 個,大多數RNA結構未知。
2019 年,史丹福大學的生物化學副教授 Rhiju Das 博士發表了一篇文章, FARFAR2: Improved de novo Rosetta prediction of complex global RNA folds 。結果顯示,在沒有實驗數據或者專家的幹預下,FARFAR2 預測的 RNA 模型更接近與 RNA 的自然構象,優於其它參與 RNA-Puzzles 的小組送出的模型。
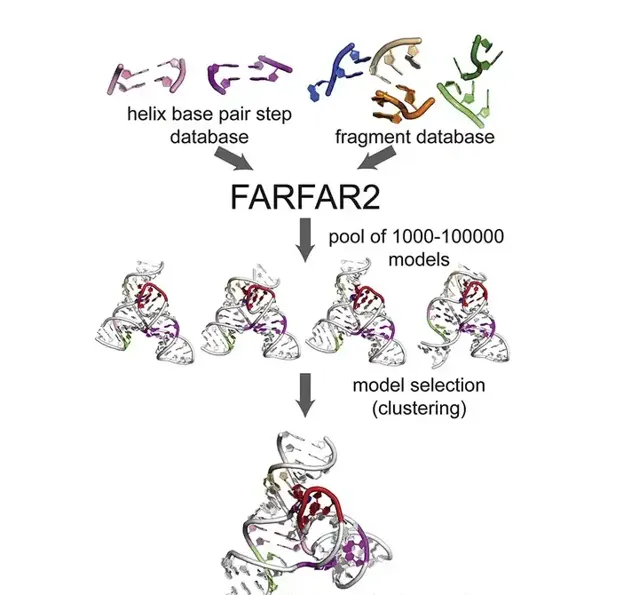
(來源: FARFAR2: Improved de novo Rosetta prediction of complex global RNA folds )
RNA-Puzzles 是由科學界組織的一項長期挑戰,旨在解析 RNA 的三級結構。根據 RNA-Puzzles 的規則,當科學家們透過實驗發現新的 RNA 結構時,他們不會公開細節,要等到其他 RNA-Puzzles 參與者送出了各自的結構預測,之後再把實驗確定的結構與這些結構進行匹配程度的對比判斷。
事實上, 研究團隊這次僅僅是為他們之前開發的 RNA 預測軟件 FARFAR2 開發了一個打分函數。
與 AI 直接預測結構不同,ARES 的框架並不是針對 RNA 的天然結構設計,而是針對人工模型與天然結構的比較,透過不斷地調整參數,ARES 可以深入了解 RNA 人工模型與天然結構上每個原子之間的相對位置以及幾何排列的不同,進而推算出人工模型與天然結構的差距有多遠。
"以前也有人做過,但從文章上來看,該模型訓練的魯棒性比較好,也就是說套用在沒見過的結構模型上,結果也不錯。 這個模型的出現對於業內的最大影響可能是,進一步讓大家認識到把深度學習套用到 RNA 結構預測上的優勢, 這也是我們組近幾年一直努力的方向。"
"但對RNA結構預測本身來說還只是一小步,因為RNA結構預測最難的部份是折疊成接近真實天然結構(near native structure)的構像,ARES 模型並沒有涉及。也就是說,ARES 模型需要跟能夠產生結構構像的 FARFAR2 結合才能預測 RNA 結構,而 FARFAR2 在多數情況下並不能產生很好的結構模型。 "周耀旗總結道。
值得註意的是,本文的通訊作者 Ron Dror 博士和 Rhiju Das 博士的研究團隊曾在 2020 年共同在 Proteins 上發表了一篇文章,利用機器學習的方式,透過生物大分子中每個原子的位置,及原子之間的相對位置,辨識蛋白質復合物的模型。本次也是繼在蛋白質上的成功,繼續探討的研究成果。
"人為因素過多,效果未知"
AlphaFold 收集了數千種已知蛋白結構進行訓練,與之不同,ARES 模型只用了 18 個已知的 RNA 結構元件,包括從 1994 年到 2006 年之間公布的數據,每個 RNA 結構元件用 Rosetta FARFAR2 生成了 1000 個結構模型。
值得註意的是,在訓練 ARES 時,並未預先將結構模型的特征編程到系統中,即使是雙螺旋、堿基對、核苷酸和氫鍵登基本概念也沒有被預先編程到系統中,而是讓演算法自己去發現。這樣做是因為如果為機器提供額外的"學習資料",會使演算法偏向於選擇某些特征,如此就會阻止它找到其他新特征。
那麽,用 18 個已知的RNA結構訓練出來的ARES模型是否可靠?
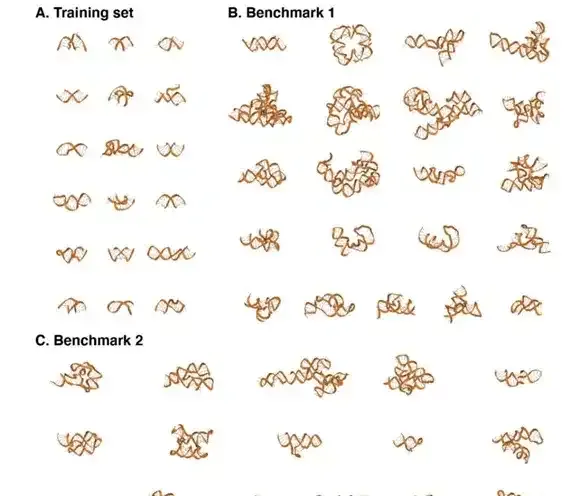
"這 18 個結構元件可以說包含了 RNA 已知結構的精要了,每個結構元件產生了 1000 個模型, 應該說 18×1000 的數據量還可以 ",周耀旗補充道,"已知的 RNA 結構很少,已知的 RNA 結構元件更少,所以這個模型能夠從 18 個結構元件訓練出來,也不完全是意外,畢竟對 ARES 模型的要求不是那麽高,把比較好的結構(更接近真實結構)的模型排序高一點,而且它是針對 FARFAR2 方法預測的模型來排序的,因為 FARFAR2 方法預測出來的模型有一定的一致性,所以訓練出來的魯棒性比較有保證。"
但周耀旗也指出, 並不能保證用 ARES 模型去給其他方法預測出來的模型打分的效果會一樣好。該模型現在僅僅是一個打分函數,裏面存在太多的人為因素,比如,研究人員選擇用 18 個 RNA 結構元件來訓練模型。
"為什麽是 18 個 RNA 結構元件來訓練模型?如果用更多的 RNA 來訓練,會有什麽不同?"周耀旗也向大家投擲了一個問題。
盡管 AlphaFold 的出現成為蛋白質結構預測領域可喜的進展,但 RNA 的結構預測依然存在著很大的問題。
蛋白質有 20 種胺基酸,包括親水、疏水、帶電荷、不帶電荷的,資訊量很大,而 RNA 只有4種堿基,資訊量少是 RNA 結構預測的難點之一。
此外,RNA 結構的穩定性低,受環境影響大,這也導致對於 RNA 結構的研究更加困難。
評分屬同類最優?
為了評估 ARES 模型的準確性,研究人員使用了從 2010 年到 2017 年間發表的 RNA 結構。對於每個 RNA 結構,研究人員使用 FARFAR2 軟件生成了至少 1,500 個結構模型。然後,他們套用經過訓練的 ARES 為每個模型生成一個分數。同時還使用了三個評分函數,分別是Rosetta、RASP、以及 3dRNAscore。
以 Rosetta 為例,在 Rosetta 中評估一個模型的好壞,最直觀的方法就是使用 Rosetta 的打分系統進行評估,也就是常說的能量函數。透過能量函數,能夠預測在什麽構象下,能量最低,RNA 處於最佳狀態,原子間的相互作用達到最優。
研究團隊用兩種方式與其它三種評分函數進行了對比。在第一個基準測試上,ARES 的效能大大優於其他三個評分功能:比如 62% 的基準 RNA 的單一最佳評分結構模型接近原生,且均高於其他三個模型。
在第二個基準測試上,ARES 再次完成了所有評分功能。而且,ARES 的最佳評分結構模型的 RNA 的中位數均方根偏差值,明顯低於其他評分函數。
研究人員稱即使在數據量少的情況下也可以成功訓練評價RNA結構的打分函數,他們希望 ARES 模型能夠幫助科學家解釋不同分子的工作原理,套用範圍從基礎生物學研究到藥物設計。
下圖可以看出,ARES 預測的 RNA 的結構模型遠接近於真實模型(4.8Å),而由 Rosetta 預測的模型則和真實模型有較大偏差( 7.7 Å )。
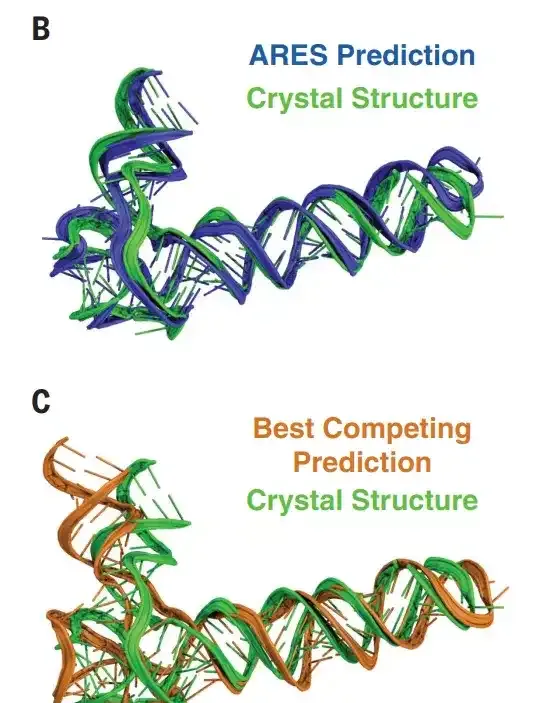
"FARFAR2 無疑是當今最好的方法之一,但目前的問題是,所有的方法都只能預測比較簡單的 RNA 結構。"周耀旗說道。
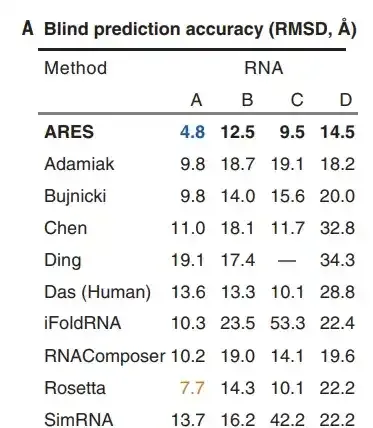
一般情況下,RNA 預測的結構至少<6 Å 才可能有點類似的模樣。事實上,在盲測比賽中,ARES+FARFAR2 所預測的 4 個 RNA,除了一個是 4.8Å,其它都是 10-15Å。看起來好像比目前的方法好很多(10-20 Å, Bujnicki組),但其實大多數誰也不像。
周耀旗總結道,ARES 其實只是相當於 AlphaFold2 裏面蛋白質人工結構評價的那部份,離 RNA 結構全預測有很大的距離。 毫無疑問,這個深度學習產生的 ARES 模型在RNA結構打分函數上邁出了可喜的一步,但這僅僅是 RNA 結構預測這個大問題裏面的一個子問題。 這個子問題和其它子問題(例如二級結構預測、主鏈構像預測和結構高精度最佳化)的一起穩定進展才能為最終解決 RNA 結構預測問題作出貢獻。
關於RNA更多相關訊息,在歷史文章中搜尋即可(gzh:生輝)。










