近日,Briefings in Bioinformatics線上發表了華中農業大學植物科學技術學院章元明團隊的題為「FastBiCmrMLM: a fast and powerful compressed variance component mixed logistic model for big genomic case-control genome-wide association study」的研究論文。該研究提出了一種多位點混合邏輯回歸全模型的大型生物樣本關聯分析高效快速FastBiCmrMLM演算法與軟件,專門檢測抗感二分類性狀或case-control數據的性狀與標記關聯,是目前執行速度很快、檢測功效最高和假陽性控制嚴格的二分類性狀關聯分析新工具。
GWAS是一種廣泛套用於動植物、微生物和人類遺傳學的基因挖掘方法。以混合線性模型為基礎的GWAS方法是在控制群體結構和遺傳背景情況下高功效檢測性狀與標記間的關聯,在過去20年得到廣泛套用和發展。然而,目前的方法存在以下問題。第一,隨著英國生物樣本庫(UK Biobank)等大型生物樣本庫的出現,關聯群體樣本量達到數十萬甚至數百萬,混合線性模型GWAS方法面臨耗時、耗運算資源的挑戰;第二,現有的大多GWAS方法通常只考慮等位基因替代效應及其遺傳背景,導致估計的效應(混雜)和控制的多基因背景均不全面,且需要假定隨機交配,降低檢測功效與精度;最後,作物抗性性狀關聯分析通常采用連續型性狀關聯分析方法,也降低了位點檢測功效與精度。為解決上述問題,本研究建立了二分類性狀的壓縮變異數組分混合邏輯回歸全模型。該模型全面考慮了所有可能效應和遺傳背景,采用章元明教授團隊已建立的壓縮變異數組分混合模型(Li et al., 2022)將四個變異數組分壓縮為兩個,顯著降低了運算復雜度,結合了一系列快速演算法和管理記憶體方法,發展了FastBiCmrMLM演算法(圖1),特別地,將SNP與性狀關聯推進至由連鎖不平衡標記構建的bin或基因單倍型與性狀關聯,為作物抗性性狀和人類復雜疾病基因挖掘提供新工具。
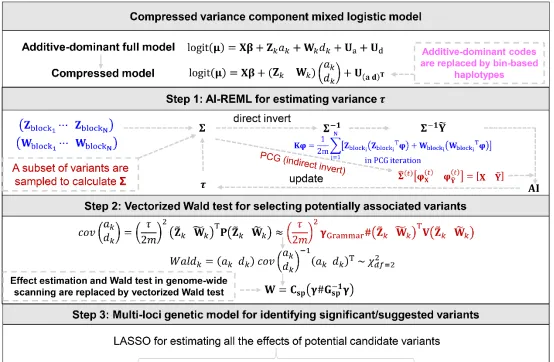
圖1. FastBiCmrMLM演算法的總體框架
FastBiCmrMLM新演算法包含了4個模組以滿足數據分析對樣本容量、運算速度、節省記憶體和功能標記的需求。分析1000個體100萬標記數據集大約需要7分鐘;分析50萬個體100萬標記大型生物樣本庫規模的數據集大約需要14小時,且可檢測大小為3‱、等位基因替代效應趨近於零和顯性效應的位點。在模擬研究中,新演算法的檢測功效比現有二分類GWAS方法更高(圖2);在5×10 -8 的顯著概率閾值下,新演算法假陽性率為4.2×10 -8 ~4.8×10 -8 ,很好地控制了假陽性率。此外,快速高效的新演算法為多組學數據分析提供新工具。新演算法的FastBiCmrMLM-Hap模組在模擬研究中可檢測到頻率為1.1%的稀有位點,在實際數據分析中能檢測到更多的稀有(<5%)位點,為稀有位點檢測提供了新思路。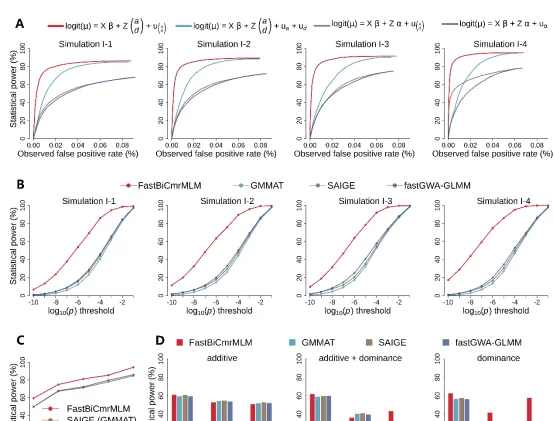
圖2. Monte Carlo模擬研究中的位點檢測功效
用FastBiCmrMLM新演算法重新分析了WTCCC數據庫((Wellcome Trust Case Control, 2007)的7個人類復雜疾病數據集。結果表明:新演算法共挖掘了29個現有方法未檢測到的有多方面證據支撐的候選基因。這一結果有力地證實了新演算法的有效性。在與這些候選基因關聯的36個顯著位點中,12個有較小的等位基因替代效應,2個只有顯性效應。這說明FastBiCmrMLM取得較高功效原因在於其更全面的模型設計,彌補了現有方法檢測小效應和顯性效應位點的不足。II型糖尿病的關聯分析與基因挖掘結果如圖3。

圖3. 二型糖尿病的顯著關聯位點及附近的候選基因
華中農業大學植物科技學院博士生王靖天為該論文第一作者,章元明教授為通訊作者,碩士研究生常曉宇和趙瓊參與了本研究。R軟件包已經在GitHub平台上釋出。本研究得到國家自然科學基金專案和惠康信托基金會(Wellcome Trust)的資助。
論文連結:https://doi.org/10.1093/bib/bbae290
參考文獻:
Wang JT, Chang XY, Zhao Q, Zhang YM. (2024) FastBiCmrMLM: a fast and powerful compressed variance component mixed logistic model for big genomic case-control genome-wide association study. Brief Bioinform 25(4): bbae290.
Li M, Zhang YW, Zhang ZC, Xiang Y, Liu MH, Zhou YH, Zuo JF, Zhang HQ, Chen Y and Zhang YM (2022) A compressed variance component mixed model for detecting QTNs and QTN-by-environment and QTN-by-QTN interactions in genome-wide association studies. MolPlant. 15: 630–650.
Wellcome Trust Case Control Consortium (2007). Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 447: 661–678.
植物科學最前沿,專註於植物科學前沿進展、 資訊、 招聘資訊的釋出及方法軟件共享等 zwkxqy ;











