更新:6/24/2020 2:10AM
谢 @Antioque 提醒,之前的回答里我发现确实只考虑了一种情况(中性底物和带负电的亲核基团)而没有继续分析其他情况,现在我在根据以往学过的基础知识加上 @Antioque 的提醒我重新梳理了一下溶剂极性对E1、E2、SN1、SN2反应的影响进行一次简单的定性分析。我简单的归纳一下,就是根据相似相溶原理, 如果反应物到过渡态活化分子的过程中极性增强,那么相应的溶剂的极性增强将加快反应速率。反之,如果反应物到过渡态活化分子的过程中极性减少,那么相应的溶剂的极性减少将加快反应速率 。极性,字面意思,部分偶极矩增大,所以极性的大小与电场强度的分布均匀性呈负相关。
补充说明:
在SN1中,底物的离去基团的离去是决速步骤,如果是中性底物 \text{R}\text{—}\text{LG} ,那么离去基团与中性底物之间的化学键断裂时电荷分布会呈现 \text{R}^{\delta+}\text{------}\text{LG}^{\delta-} ,由于碳正离子 \text{R}^{+} 的点正电荷增大并且离去基团 \text{LG} 的点负电荷也增大,导致电场强度分布更不均匀,即极性增大。如果是带正电的底物 \text{R}\text{—}\text{LG}^{+} ,那么离去基团与中性底物之间的化学键断裂时电荷分布会呈现 \text{R}^{\delta+}\text{------}\text{LG}^{\delta+} ,由于碳正离子 \text{R}^{+} 的点正电荷增大而离去基团 \text{LG} 的点正电荷减弱,导致电场强度分布更均匀,即极性减小。而极性溶剂中质子化溶剂又比非质子化溶剂极性更强,使得中性底物的解离能显著性降低,如果底物比作一张纸,那么溶剂的极性大小就是双手撕扯纸张的强弱。例如水 \text{H}_{2}\text{O} 可以微弱自解离成 \text{H}_{3}\text{O}^{+} 和 \text{OH}^{-} ,然后诱导效应异性相吸使得反应物从初态 \text{OH}^{-}\text{------}\text{R}\text{—}\text{LG}\text{------}\text{H}_{3}\text{O}^{+} 到过渡态 \text{OH}^{-}\text{---}\text{R}^{\delta+}\text{------}\text{LG}^{\delta-}\text{---}\text{H}_{3}\text{O}^{+} 相比于非质子化和非极性溶剂所需的解离能更低。对于带正电的底物 \text{R}\text{—}\text{LG}^{+} 则相反,由于极性反转,即反应物初态变成 \text{H}_{3}\text{O}^{+}\text{------}\text{R}\text{—}\text{LG}^{+}\text{------}\text{OH}^{-} 根据异性相吸原理,导致过渡态 \text{H}_{3}\text{O}^{+}\text{---}\text{R}^{\delta+}\text{------}\text{LG}^{\delta+}\text{---} \text{OH}^{-} 中的电场分布相比于初态更不均匀,即更不稳定。所需的活化能也就相比于弱极性或非极性溶剂环境下更高。如果把底物比作被压缩的弹簧,那么极性分子就是那双摁住弹簧的手。
在SN2中,由于在动力学下是一步反应,所以亲核基团和底物的活性共同决定反应速率,根据电性一共有2*2=4种情况
- 中性底物和负电性亲核基团 \text{Nu}^{-}\text{------}\text{R}\text{—}\text{LG}\rightarrow \text{Nu}^{\delta-}\text{---}\text{R}\text{------}\text{LG}^{\delta-}
- 中性底物和中性亲核基团 \text{Nu}\text{------}\text{R}\text{—}\text{LG}\rightarrow \text{Nu}^{\delta+}\text{---}\text{R}\text{------}\text{LG}^{\delta-}
- 正电性底物和负电性亲核基团 \text{Nu}^{-}\text{------}\text{R}\text{—}\text{LG}^{+}\rightarrow \text{Nu}^{\delta-}\text{---}\text{R}\text{------}\text{LG}^{\delta+}
- 正电性底物和中性亲核基团 \text{Nu}\text{------}\text{R}\text{—}\text{LG}^{+}\rightarrow \text{Nu}^{\delta+}\text{---}\text{R}\text{------}\text{LG}^{\delta+}
第一、三和四种情况里电场强度分布更均匀,即极性减少。而第二种则相反,电场强度分布更不均匀,即极性增大。所以对于第一、三、四种情况,减少溶剂的极性有助于加快反应速率。对于第二种情况,增加溶剂的极性有助于加快反应速率。
E1和E2与SN1和SN2基本同理,其中有区别的地方是E2和SN2中使用的溶剂和亲核试剂(Lewis碱)的相互作用会影响Lewis碱的亲核性。质子化溶剂均会抑制中性和负电性亲核试剂的亲核性,因为亲核试剂作为Lewis碱,即给电子体会使得该亲核试剂的偏负电部分会与溶剂的偏正电部分——氢原子形成氢键,导致亲核试剂与质子化溶剂配合或因高位阻效应而失去亲核性,所以抑制了SN2反应。当然也会抑制SN1反应但是相比SN2,SN1反应要求的Lewis碱不需要那么强的亲核性。温度也是一个重要的因素,通常来讲消去反应相对高温。
以上就是现阶段我已经回顾的大二有机化学一部分基础知识了,如果有什么遗漏、错误和不严谨的地方也请各位读者在评论区留言。最近本人在专注于数学和计算机选修,以便以后跨专业领域学习与工作。
原回答
题主首先得了解这四种反应的机制才能了解具体在什么条件下哪种反应占主导(酸碱性、极性、温度、位阻效应、活化配合物稳定性、手性等等等等)。我看了题目的简介,题主是想着重分析溶剂极性的影响,那么此回答我就着重分析极性,当然要记住还有其他因素会影响这四种反应之间的竞争。以下是我对大二时期学的有机化学知识的回顾。总的来说
E1是单分子消去反应,先是底物中的离去基团被解离并且在原中心碳位置上形成碳正离子,然后邻位β碳原子上的氢被拔去后形成π键,该反应在动力学上分为两部,第一步是速率决定步骤,所以底物的活性影响反应速率。
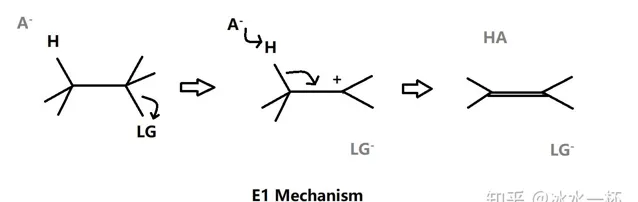
该反应在极性溶剂条件下更有利,这是因为离去基团的解离并形成碳正离子是决速步骤,形成的碳正离子会与极性溶剂分子的偏负电荷部分形成离子偶极矩分散电荷密度从而保持稳定(就像食盐水溶液中的钠离子和水分子中的偏负电荷部分——氧原子上的相互作用),即溶剂化效应。质子极性溶剂稳定作用更强,那是因为形成的离子偶极矩是氢键,氢键比大多数分子间相互作用强。所以该反应非常适合用酸催化。因为在第二步sp3杂化碳原子上的C-H键断裂时需要相应的活化能,所以E1反应通常在相对高温的环境下进行,否则会发生SN1反应而不是E1反应(详细见下文SN1反应)。同时,温度的上升也会加强溶剂化效应,这是因为温度的上升使得溶剂分子的自解离反应平衡向解离方向移动从而增强溶剂的极性。
E2是双分子消去反应,Lewis碱直接进攻拔去邻位β碳原子上的氢同时对位上的离去基团被解离从而形成π键,该反应在动力学上只有一步,底物和Lewis碱的活性共同影响反应速率

该反应在非极性溶剂条件下更有利,这是因为Lewis碱中的偏负电荷部分会与极性溶剂分子的偏正电荷部分形成离子偶极矩分散电荷密度从而保持稳定(就像食盐水溶液中的氯离子和水分子中的偏正电荷部分——氢原子上的相互作用),即溶剂化效应。质子极性溶剂分子甚至会直接与Lewis碱配合从而失去反应活性。所以该反应的发生需要强Lewis碱和高位阻效应(详细见下文SN2反应)同时,温度的上升也会加强溶剂化效应,从而在热力学上减弱亲核基团的亲核性并且增强邻位碳上的氢的解离性(达到解离所需的活化能)。所以在实验室里一般E2反应是在相对高温的环境下进行。
SN1是单分子取代反应,先是底物中的离去基团被解离并且在原中心碳位置上形成碳正离子,然后亲核基团进攻碳正离子形成新的σ键,该反应在动力学上分为两部,第一步是速率决定步骤,所以底物的活性影响反应速率。
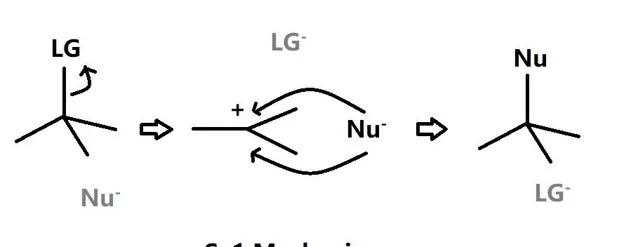
由此可以看出,与E1反应类似,该反应在极性溶剂条件下更有利,但是要注意,温度的上升也会促进生成产物中的亲核基团的解离性,从而在热力学上降低生成产物的稳定性从而可能发生E1反应而不是SN1反应,即存在E1和SN1反应的天然竞争。
回顾下乙醇在浓硫酸的催化下脱水生成乙烯的E1反应 \text{EtOH}\xrightarrow[170°\text{C}]{\text{H}_{2}\text{SO}_{4}} \text{C}_{2}\text{H}_{4}+\text{H}_{2}\text{O} 中的机理,第一步是决速步骤,即乙醇在酸性条件下脱水形成乙基正离子 \text{EtOH}+\text{H}^{+}\rightarrow\text{C}_{2}\text{H}_{5}^{+}+\text{H}_{2}\text{O} ,但是在第二步中,如果反应条件是在常温条件下,即不满足170度的高温,那么形成的碳正离子会直接被亲核基团硫酸氢根离子进攻从而发生SN1反应并形成硫酸氢乙酯(单乙基硫酸) \text{C}_{2}\text{H}_{5}^{+}+\text{H}\text{SO}_{4}^{-}\rightarrow \text{EtHSO}_{4} 。 随着温度的上升,硫酸氢乙酯开始解离,在140度左右,由于在热力学上没有达到解离乙基正离子中sp3杂化的碳上的氢所需的活化能,另一个乙醇分子上的氧上的一对孤电子会进攻已经解离出的乙基正离子然后被硫酸氢根离子拔去额外的氢从而生成乙醚 \text{EtOH}+\text{C}_{2}\text{H}_{5}^{+}+\text{HSO}_{4}^{-}\rightarrow \text{EtOEt}+\text{H}_{2}\text{SO}_{4} 。在170度时才会发生E1反应,乙基正离子上的邻位碳上的氢被拔去并形成π键从而生成乙烯 \text{C}_{2}\text{H}_{5}^{+}+\text{HSO}_{4}^{-}\rightarrow \text{C}_{2}\text{H}_{4}+\text{H}_{2}\text{SO}_{4} 。
SN2是双分子取代反应,亲核基团直接进攻偏正电荷的碳同时对位上的离去基团被解离从而形成新的σ键,该反应在动力学上只有一步,底物和亲核基团的活性共同影响反应速率。
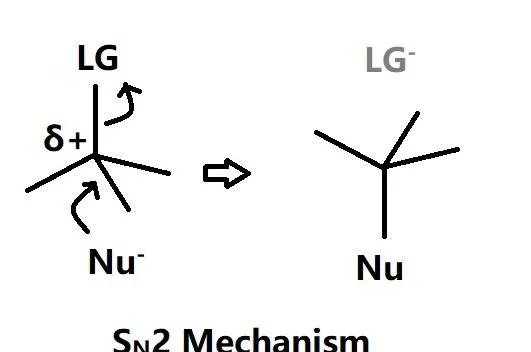
与E2反应类似,该反应在非极性溶剂条件下更有利,该反应的发生需要强Lewis碱和低位阻效应(例如小分子强亲核性Lewis碱乙氧基负离子 \text{EtO}^{-} )。如果是大分子弱亲核性Lewis碱如叔丁氧基负离子 t\text{BuO}^{-} (叔丁醇 (\text{CH}_{3})_{3}\text{COH} 脱去羟基上的氢形成) 那么就可能会发生E2反应,即Lewis碱不会进攻偏正电荷的碳而是拔去邻位碳上的氢。即存在E2和SN2反应的天然竞争。同时,温度的上升也会加强溶剂化效应,从而在热力学上减弱亲核基团的亲核性并且增强邻位碳上的氢的解离性(达到解离所需的活化能)。所以在实验室里一般SN2反应是在相对低温的环境下进行。
总结
极性低温——SN1
极性高温——E1
非极性低温——SN2
非极性高温——E2
要记住,还有其他因素(区域选择性,手性,共振结构等等等等)会影响这四种反应的竞争,这个总结是在其他因素等同的条件下说明的。




